You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5795)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Mitochondrial Disease FactsheetDocument3 pagesMitochondrial Disease FactsheetNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Foreign Affairs May June 2021 IssueDocument216 pagesForeign Affairs May June 2021 IssueSohail BhattiNo ratings yet
- English Idioms and PhrasesDocument384 pagesEnglish Idioms and Phrasesthwe thweNo ratings yet
- Heroic Tales Core Rules 1.1.0Document33 pagesHeroic Tales Core Rules 1.1.0Melobajoya MelobajoyaNo ratings yet
- 06 Renr5908 08 01 All PDFDocument108 pages06 Renr5908 08 01 All PDFFrancisco Ospino Arrieta100% (2)
- Koehring ManualDocument56 pagesKoehring ManualKyle A. Nolan100% (3)
- Mooka Panchsati Arya SatakamDocument18 pagesMooka Panchsati Arya SatakamPrasad Raviprolu100% (1)
- T.Y.B.B.A. (CA) Sem VI Practical Slips 2019 PatternDocument30 pagesT.Y.B.B.A. (CA) Sem VI Practical Slips 2019 PatternJai Ramteke100% (2)
- Contingency Measures and ProceduresDocument25 pagesContingency Measures and ProceduresKaren Villapando LatNo ratings yet
- Apc 10kva Ups ManualDocument36 pagesApc 10kva Ups Manualraj rajNo ratings yet
- Alpers-Huttenlocher FactsheetDocument2 pagesAlpers-Huttenlocher FactsheetNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Session Four: HomeworkDocument1 pageSession Four: HomeworkNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Family History CollectionDocument1 pageFamily History CollectionNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Genomics For The Child Neurologist Facilitator Guide Session Four Results Intepretation and ApplicationDocument7 pagesGenomics For The Child Neurologist Facilitator Guide Session Four Results Intepretation and ApplicationNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Results Interpretation & Application Toolkit: ManagementDocument1 pageResults Interpretation & Application Toolkit: ManagementNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Informed Consent Talking PointsDocument4 pagesInformed Consent Talking PointsNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Genetic Testing Process Homework: Published June 2013 © Nchpeg All Rights ReservedDocument1 pageGenetic Testing Process Homework: Published June 2013 © Nchpeg All Rights ReservedNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Family History CollectionDocument1 pageFamily History CollectionNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Informed Consent ChecklistDocument2 pagesInformed Consent ChecklistNational Coalition for Health Professional Education in GeneticsNo ratings yet
- CMA Provider ResourcesDocument1 pageCMA Provider ResourcesNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Dravet Syndrome FactsheetDocument1 pageDravet Syndrome FactsheetNational Coalition for Health Professional Education in GeneticsNo ratings yet
- HBOC Red Flags ChecklistDocument1 pageHBOC Red Flags ChecklistNational Coalition for Health Professional Education in GeneticsNo ratings yet
- HBOC Family History Collection ToolDocument4 pagesHBOC Family History Collection ToolNational Coalition for Health Professional Education in GeneticsNo ratings yet
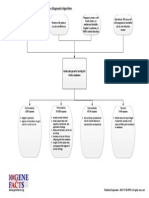
- Fragile X Syndrome Diagnostic AlgorithmDocument1 pageFragile X Syndrome Diagnostic AlgorithmNational Coalition for Health Professional Education in GeneticsNo ratings yet
- Have Been Tried From Time To Time," As Churchill Famously Said (Third Paragraph) "?Document25 pagesHave Been Tried From Time To Time," As Churchill Famously Said (Third Paragraph) "?Aditya ThakurNo ratings yet
- Interdisciplinary Project 1Document11 pagesInterdisciplinary Project 1api-424250570No ratings yet
- Unit 9 TelephoningDocument14 pagesUnit 9 TelephoningDaniela DanilovNo ratings yet
- Asia Pacific SAR Plan V2.0Document38 pagesAsia Pacific SAR Plan V2.0Joci SimõesNo ratings yet
- Bearing Repeater CompassDocument4 pagesBearing Repeater CompassJohn PerdyNo ratings yet
- Operating Instructions: Vacuum Drying Oven Pump ModuleDocument56 pagesOperating Instructions: Vacuum Drying Oven Pump ModuleSarah NeoSkyrerNo ratings yet
- ĐÁP ÁN ĐỀ THI THỬ SỐ 03 (2019-2020)Document8 pagesĐÁP ÁN ĐỀ THI THỬ SỐ 03 (2019-2020)Đào VânNo ratings yet
- A. Erfurth, P. Hoff. Mad Scenes in Early 19th-Century Opera PDFDocument4 pagesA. Erfurth, P. Hoff. Mad Scenes in Early 19th-Century Opera PDFbiarrodNo ratings yet
- January 2014 QP - Paper 1 Edexcel (B) Maths IGCSEDocument24 pagesJanuary 2014 QP - Paper 1 Edexcel (B) Maths IGCSEStevenstrange001 CattyNo ratings yet
- MikroekonomiDocument1 pageMikroekonomiYudhaPrakosoIINo ratings yet
- Manipulation Methods and How To Avoid From ManipulationDocument5 pagesManipulation Methods and How To Avoid From ManipulationEylül ErgünNo ratings yet
- Class XI-Writing-Job ApplicationDocument13 pagesClass XI-Writing-Job Applicationisnprincipal2020No ratings yet
- Variables in The EquationDocument3 pagesVariables in The EquationfiharjatinNo ratings yet
- CE - 441 - Environmental Engineering II Lecture # 11 11-Nov-106, IEER, UET LahoreDocument8 pagesCE - 441 - Environmental Engineering II Lecture # 11 11-Nov-106, IEER, UET LahoreWasif RiazNo ratings yet
- Lesson 3: Letters of RequestDocument4 pagesLesson 3: Letters of RequestMinh HiếuNo ratings yet
- 311762en WDocument36 pages311762en WOprisor CostinNo ratings yet
- Warranties Liabilities Patents Bids and InsuranceDocument39 pagesWarranties Liabilities Patents Bids and InsuranceIVAN JOHN BITONNo ratings yet
- Practical Econometrics Data Collection Analysis and Application 1st Edition Hilmer Test BankDocument27 pagesPractical Econometrics Data Collection Analysis and Application 1st Edition Hilmer Test Bankdavidhallwopkseimgc100% (28)
- Declaration of Absence of Conflict of InterestDocument1 pageDeclaration of Absence of Conflict of InterestJvhelcoronacondat CondatNo ratings yet
- DR Cast Iron Fittings CharlotteDocument124 pagesDR Cast Iron Fittings CharlotteMohamad NohayliNo ratings yet
- Paper 19 AugustDocument552 pagesPaper 19 AugustUma Sankar Pradhan100% (1)