You might also like
- Anesthesia NotesDocument9 pagesAnesthesia NotesezNo ratings yet
- Ampalaya Chips With Sweet DipDocument31 pagesAmpalaya Chips With Sweet DipJayson San Juan86% (7)
- Cell Signaling IDocument21 pagesCell Signaling IGuhanNo ratings yet
- Cellular and Molecular Mechanisms of Inflammation: Receptors of Inflammatory Cells: Structure—Function RelationshipsFrom EverandCellular and Molecular Mechanisms of Inflammation: Receptors of Inflammatory Cells: Structure—Function RelationshipsCharles G. CochraneNo ratings yet
- Burden of Psoriasis Report FinalDocument52 pagesBurden of Psoriasis Report FinalmeilanydianiNo ratings yet
- My Journal 2Document11 pagesMy Journal 2FelixNo ratings yet
- Research Article: Metformin Induces Cell Cycle Arrest and Apoptosis in Drug-Resistant Leukemia CellsDocument12 pagesResearch Article: Metformin Induces Cell Cycle Arrest and Apoptosis in Drug-Resistant Leukemia Cellsnurfitriani runduNo ratings yet
- Microbiota or Short-Chain Fatty Acids: Which Regulates Diabetes?Document4 pagesMicrobiota or Short-Chain Fatty Acids: Which Regulates Diabetes?fransiscuNo ratings yet
- tmp3AAA TMPDocument10 pagestmp3AAA TMPFrontiersNo ratings yet
- MainDocument8 pagesMainAry MadinaNo ratings yet
- KIT and PDGFRA Mutations in GISTs Guide TreatmentDocument22 pagesKIT and PDGFRA Mutations in GISTs Guide TreatmentCarla CarolineNo ratings yet
- Regulation of Protein and MRNA Expression of The MTORC1 RepressorDocument6 pagesRegulation of Protein and MRNA Expression of The MTORC1 RepressorIsa AguilarNo ratings yet
- P-glycoprotein Structure and FunctionDocument5 pagesP-glycoprotein Structure and FunctionZiedTrikiNo ratings yet
- 339 FullDocument8 pages339 FullKay BristolNo ratings yet
- Cyclin Dependent Kinase As AnticancerDocument18 pagesCyclin Dependent Kinase As AnticancerLydia Angelia YanitaNo ratings yet
- Epigenetic Regulation of Cancer-Associated Gene ProductsDocument27 pagesEpigenetic Regulation of Cancer-Associated Gene ProductsPace RaditNo ratings yet
- Growth Factors and Growth Factor Receptors in Cancer: T. RajkumarDocument7 pagesGrowth Factors and Growth Factor Receptors in Cancer: T. RajkumarMohammed AladhraeiNo ratings yet
- p38 Mitogen-Activated Protein Kinase Is The Central Regulator of Cyclic AMP-Dependent Transcription of The Brown Fat Uncoupling Protein 1 GeneDocument11 pagesp38 Mitogen-Activated Protein Kinase Is The Central Regulator of Cyclic AMP-Dependent Transcription of The Brown Fat Uncoupling Protein 1 GeneAlmir FilsNo ratings yet
- 1 s2.0 S1567576918303606 MainDocument10 pages1 s2.0 S1567576918303606 Mainsicongli.leonleeNo ratings yet
- Site-Dependent Differential KIT and PDGFRA Expression in Gastric and Intestinal Gastrointestinal Stromal TumorsDocument10 pagesSite-Dependent Differential KIT and PDGFRA Expression in Gastric and Intestinal Gastrointestinal Stromal TumorsElisa KartikaNo ratings yet
- Mendel Et Al 2003Document12 pagesMendel Et Al 2003Selliana Maretha Wijaya KusumaNo ratings yet
- Alvarado Lahip2Document11 pagesAlvarado Lahip2api-559892740No ratings yet
- PIIS1535610819302971Document11 pagesPIIS1535610819302971saraanpkNo ratings yet
- Expresion Patterns of Genes, Regulating Lipid Metabolism in Prostate TumorsDocument16 pagesExpresion Patterns of Genes, Regulating Lipid Metabolism in Prostate TumorsАнна ШаповаловаNo ratings yet
- J. Biol. Chem.-1999-Cotrim-37723-30Document8 pagesJ. Biol. Chem.-1999-Cotrim-37723-30JudeMucaNo ratings yet
- Impact of Dietary Components On NK and Treg Cell Function For Cancer PreventionDocument6 pagesImpact of Dietary Components On NK and Treg Cell Function For Cancer PreventionEgy SunandaNo ratings yet
- Pro Metastatic Signaling of The Trans Fatty Acid Elaidic Acid Is Associated With Lipid RaftsDocument4 pagesPro Metastatic Signaling of The Trans Fatty Acid Elaidic Acid Is Associated With Lipid RaftsKaren Andrea StNo ratings yet
- Adaptor Protein-2 Interaction With Arrestin Regulates GPCR Recycling and ApoptosisDocument15 pagesAdaptor Protein-2 Interaction With Arrestin Regulates GPCR Recycling and ApoptosisBruna FrancieleNo ratings yet
- Molecular CancerDocument25 pagesMolecular CancergpaivNo ratings yet
- Lipoic Acid Inhibits Cell Proliferation of Tumor Cells in Vitro and in VivoDocument11 pagesLipoic Acid Inhibits Cell Proliferation of Tumor Cells in Vitro and in VivosweetohmNo ratings yet
- KongDocument6 pagesKongapi-390257815No ratings yet
- Sciencedirect: ArticleinfoDocument7 pagesSciencedirect: ArticleinfoSergeat18BNo ratings yet
- R49 Full PDFDocument14 pagesR49 Full PDFJuan Martin Perez GrilloNo ratings yet
- The Roles of Cyclin-Dependent Kinases in Cell-CyclDocument28 pagesThe Roles of Cyclin-Dependent Kinases in Cell-CyclcekanajsenaNo ratings yet
- FC AntybodyDocument8 pagesFC AntybodyahmaszitimNo ratings yet
- Article: Correspondence: Lrobinso@uoguelph - Ca Tel.: +1-519-824-4120 (Ext. 52297)Document16 pagesArticle: Correspondence: Lrobinso@uoguelph - Ca Tel.: +1-519-824-4120 (Ext. 52297)Alma AcevedoNo ratings yet
- The PI3K Akt Pathway Is One of The Most Important Survival Signaling Cascades Altered in Human Solid Tumors Including Pros Tate CancerDocument2 pagesThe PI3K Akt Pathway Is One of The Most Important Survival Signaling Cascades Altered in Human Solid Tumors Including Pros Tate Cancerpumahell25No ratings yet
- A Chemical Biology Screen Identifies Glucocorticoids That Regulate C - 2007 - BLDocument8 pagesA Chemical Biology Screen Identifies Glucocorticoids That Regulate C - 2007 - BLTareeqanwar MohammedNo ratings yet
- 15G Protein-Coupled ReceptorDocument15 pages15G Protein-Coupled ReceptorZiedTrikiNo ratings yet
- Articulo de Investigacion 1Document20 pagesArticulo de Investigacion 1HÉCTOR MEDINANo ratings yet
- Arginine Deiminase As A Novel Therapy For Prostate Cancer Induces Autophagy and Caspase-Independent ApoptosisDocument9 pagesArginine Deiminase As A Novel Therapy For Prostate Cancer Induces Autophagy and Caspase-Independent ApoptosisAvishekh SinhaNo ratings yet
- Molecular Biology of Pharmacology and GeneticsDocument39 pagesMolecular Biology of Pharmacology and GeneticsSusi RutmalemNo ratings yet
- Glucagon Regulates Gluconeogenesis Through KAT2B-and WDR5-mediated Epigenetic EffectsDocument11 pagesGlucagon Regulates Gluconeogenesis Through KAT2B-and WDR5-mediated Epigenetic EffectsLorenaNo ratings yet
- Regulation of Apoa Gene Expression With Acidosis: Requirement For A Transcriptional RepressorDocument15 pagesRegulation of Apoa Gene Expression With Acidosis: Requirement For A Transcriptional RepressorAbdur Rachman Ba'abdullahNo ratings yet
- Epigenetic Landscape in Blood Leukocytes Following Ketosis and - 2021 - ClinicaDocument14 pagesEpigenetic Landscape in Blood Leukocytes Following Ketosis and - 2021 - ClinicaArie Dwi ANo ratings yet
- CDD Is 2013151 ADocument10 pagesCDD Is 2013151 ADalton JLNo ratings yet
- Gpcrs and CancervistaDocument45 pagesGpcrs and CancervistapriyaaNo ratings yet
- CDD 2011102 ADocument12 pagesCDD 2011102 ACaerulus Fuad Abdul BaqiNo ratings yet
- GPT2 - Gls1 - Sensitivity - 2020Document12 pagesGPT2 - Gls1 - Sensitivity - 2020shravanvvNo ratings yet
- Chen S - 2017Document10 pagesChen S - 2017Tiago OgaitNo ratings yet
- Dorseuil Et Al 1992 - Inhibition of Superoxide Production in B Lymphocytes by Rac Antisense OligonucleotidesDocument3 pagesDorseuil Et Al 1992 - Inhibition of Superoxide Production in B Lymphocytes by Rac Antisense OligonucleotidesHernestoNo ratings yet
- 12 04aoncataboliterepressionDocument11 pages12 04aoncataboliterepressionMella Rosa SebiferaNo ratings yet
- Probiotic Lactobacillus and Estrogen Effects On Vaginal Epithelial Gene Expression Responses To Candida AlbicansDocument10 pagesProbiotic Lactobacillus and Estrogen Effects On Vaginal Epithelial Gene Expression Responses To Candida AlbicansYadi PurnomoNo ratings yet
- Tumor Cell Pseudopodial ProtrusionsDocument10 pagesTumor Cell Pseudopodial Protrusionsvanhuynh2812No ratings yet
- Dedifferentiation of podocytes in diabetes modelDocument10 pagesDedifferentiation of podocytes in diabetes modelEktho11No ratings yet
- Helicobacter Pylori For The Synthesis of Nucleotide SugarsDocument7 pagesHelicobacter Pylori For The Synthesis of Nucleotide SugarsSUNIL KUMAR JANGRANo ratings yet
- Role of Galectins in Inflammatory and Immunomodulatory Processes 2002Document11 pagesRole of Galectins in Inflammatory and Immunomodulatory Processes 2002evilbioNo ratings yet
- Edit Nutrigenomik DAS KLP 1Document37 pagesEdit Nutrigenomik DAS KLP 1Grace KaunangNo ratings yet
- Project ProposalDocument8 pagesProject ProposalArnab ChakrabortyNo ratings yet
- Boumediene Bouzahzah - 2001 - 274 PDFDocument15 pagesBoumediene Bouzahzah - 2001 - 274 PDFarnipahlawaniNo ratings yet
- Archives of Biochemistry and Biophysics: SciencedirectDocument6 pagesArchives of Biochemistry and Biophysics: SciencedirectAlyna AlynaNo ratings yet
- Molecular Diagnosis of Type 1c Glycogen Storage DDocument1 pageMolecular Diagnosis of Type 1c Glycogen Storage DDeeptika Agarwal YadavNo ratings yet
- Seminario 4A Regulacion de La GlicemiaDocument6 pagesSeminario 4A Regulacion de La GlicemiaEdol LopezNo ratings yet
- List CartoonDocument5 pagesList CartoonNadia Novia WulandariNo ratings yet
- List CartoonDocument5 pagesList CartoonNadia Novia WulandariNo ratings yet
- 169 FullDocument12 pages169 FullNadia Novia WulandariNo ratings yet
- Hormonal Control of Energy and Protein Metabolism: G. H. Mcdowell and E. F. AnnisonDocument1 pageHormonal Control of Energy and Protein Metabolism: G. H. Mcdowell and E. F. AnnisonNadia Novia WulandariNo ratings yet
- 169 FullDocument12 pages169 FullNadia Novia WulandariNo ratings yet
- Effects of Coadministration of Extract of Carica Hypoglycemic AgentsDocument8 pagesEffects of Coadministration of Extract of Carica Hypoglycemic AgentsNeri MargaretNo ratings yet
- Glucocorticoid-Resistant Asthma: Pathogenesis and Clinical Implications For ManagementDocument8 pagesGlucocorticoid-Resistant Asthma: Pathogenesis and Clinical Implications For ManagementNadia Novia WulandariNo ratings yet
- 1367 FullDocument7 pages1367 FullNadia Novia WulandariNo ratings yet
- Justus Von LiebigDocument7 pagesJustus Von LiebigNadia Novia WulandariNo ratings yet
- Law of ToleranceDocument2 pagesLaw of ToleranceAhmed RashidNo ratings yet
- Tenebrio MolitorDocument9 pagesTenebrio Molitorじょしら フィアンナNo ratings yet
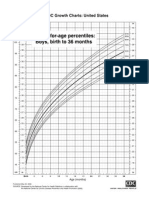
- CDC Boys 0 - 36 Mths - Length-For-AgeDocument1 pageCDC Boys 0 - 36 Mths - Length-For-AgepmverlaanNo ratings yet
- Main PDFDocument3 pagesMain PDFAlmira RahmaNo ratings yet
- Customizing Anaphylaxis Guidelines For Emergency MedicineDocument8 pagesCustomizing Anaphylaxis Guidelines For Emergency MedicineCAMILA FERNANDA ROMERO GODOYNo ratings yet
- Dc81b Colour Out of Space Ver 001Document3 pagesDc81b Colour Out of Space Ver 001Victor Guido PinheiroNo ratings yet
- PHD Thesis Public Health PDFDocument7 pagesPHD Thesis Public Health PDFangelicaortizpaterson100% (2)
- Principles of Sterile Field and Definitions or Romm PresentationDocument29 pagesPrinciples of Sterile Field and Definitions or Romm PresentationAprille Claire MoralesNo ratings yet
- 001 210197064 Ci2 117 1Document1 page001 210197064 Ci2 117 1tooba arshadNo ratings yet
- Second Announcement JASS RevisiDocument28 pagesSecond Announcement JASS RevisiPoco BlueNo ratings yet
- Lecture - 1 Dosage FormDocument13 pagesLecture - 1 Dosage FormAshique Farhad100% (1)
- Short Cases in MedicineDocument30 pagesShort Cases in MedicineselamuNo ratings yet
- Medicine Cupboard Keys Part BDocument6 pagesMedicine Cupboard Keys Part BSojiNo ratings yet
- Routine Medical Checkup PublicationDocument11 pagesRoutine Medical Checkup Publication백만호No ratings yet
- The Human Body Frequency: March 2020Document3 pagesThe Human Body Frequency: March 2020Dejya CordoraNo ratings yet
- Inmuno 555Document11 pagesInmuno 555Daniela Fdz CNo ratings yet
- Preterm LaborDocument29 pagesPreterm LaborBer AnneNo ratings yet
- C1 C2 E1 E2Document11 pagesC1 C2 E1 E2araly83No ratings yet
- Asada 2Document33 pagesAsada 2Anonymous iNLNBVtpoNo ratings yet
- Scott-Brown's Vol 2 Benign Neck Disease - Infections & SwellingDocument12 pagesScott-Brown's Vol 2 Benign Neck Disease - Infections & SwellingjessNo ratings yet
- DrugsDocument1 pageDrugsMarnelie Guerrero AbuanNo ratings yet
- Novel Anti-Inflammatory Treatments For Asthma: ReviewDocument13 pagesNovel Anti-Inflammatory Treatments For Asthma: Reviewnopera kurnia watiNo ratings yet
- NCP Con ConstipationDocument2 pagesNCP Con ConstipationChristine Marie Bucio OraizNo ratings yet
- 66Document82 pages66Ankit kumar singhNo ratings yet
- BioPsych Lecture Notes CH 7Document6 pagesBioPsych Lecture Notes CH 7Generic_Persona100% (1)
- Minor Scrub 3 PDFDocument3 pagesMinor Scrub 3 PDFMadel AcobNo ratings yet
- Gestational Hypertension and Preeclampsia ACOG Practice Bulletin, Number 222 1605448006Document24 pagesGestational Hypertension and Preeclampsia ACOG Practice Bulletin, Number 222 1605448006xibalba.edNo ratings yet
- Thesis OphthalmologyDocument7 pagesThesis OphthalmologyJames Heller100% (2)