You might also like
- Internal Combustion EngineDocument20 pagesInternal Combustion EngineRowin Mark Sabornido33% (3)
- Thermodynamic Lesson 3Document5 pagesThermodynamic Lesson 3kelebekkNo ratings yet
- E Rathakrishnan Gas Dynamics SolutionsDocument216 pagesE Rathakrishnan Gas Dynamics SolutionsVigneshVickey67% (15)
- Physics - Solution Set For Homework Book Chapter - 15 KTG & ThermodynamicsDocument4 pagesPhysics - Solution Set For Homework Book Chapter - 15 KTG & ThermodynamicsKishorNo ratings yet
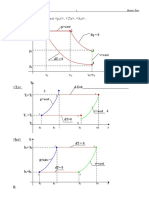
- Ideal Gas ProcessesDocument6 pagesIdeal Gas ProcessesKlydeJoseNo ratings yet
- Isothermal Gas Expansion: P V T P V TDocument4 pagesIsothermal Gas Expansion: P V T P V Twolfofphysics08IPMP01No ratings yet
- 351 F 22 Exam EquationsDocument1 page351 F 22 Exam EquationsEdaNo ratings yet
- Formulario Termodinamica-2Document1 pageFormulario Termodinamica-2Anonymous 2x6VE65kNo ratings yet
- CARNOT CYCLE EFFICIENCY AND MEAN PRESSUREDocument6 pagesCARNOT CYCLE EFFICIENCY AND MEAN PRESSUREclarkmaxNo ratings yet
- State Equations ExplainedDocument98 pagesState Equations ExplainedAnonymous xmSWrWbUKGNo ratings yet
- Termo 1' Modeli 3.doc EraldiDocument12 pagesTermo 1' Modeli 3.doc EraldiFeritNo ratings yet
- Solution Paper - 02: 199 200 Cos 2 Cos CosDocument7 pagesSolution Paper - 02: 199 200 Cos 2 Cos CosLokesh KumarNo ratings yet
- Chapter 3 - Section B - Non-Numerical SolutionsDocument12 pagesChapter 3 - Section B - Non-Numerical SolutionsAwaludin R FirmanshahNo ratings yet
- Problems303 2 SolDocument3 pagesProblems303 2 SolElaineNo ratings yet
- CHE3161 Semester1 2010 Solutions PDFDocument14 pagesCHE3161 Semester1 2010 Solutions PDFkumiristineNo ratings yet
- Sasi Institute of Technology EnineeringDocument6 pagesSasi Institute of Technology EnineeringHamu NalaNo ratings yet
- Adiabatic ProcessDocument4 pagesAdiabatic ProcessDamodaram MadaliNo ratings yet
- Formulario Termodinamica 1Document2 pagesFormulario Termodinamica 1Jesus LyroyNo ratings yet
- Pressure and Thermodynamics FundamentalsDocument2 pagesPressure and Thermodynamics FundamentalsPearl Alexandra FabitoNo ratings yet
- Physics 112 formulae cheat sheetDocument3 pagesPhysics 112 formulae cheat sheetSaied RajehaNo ratings yet
- Physics Olympiad Preparation Program 2010-2011: - University of TorontoDocument10 pagesPhysics Olympiad Preparation Program 2010-2011: - University of TorontoAlejandro Garcia PerezNo ratings yet
- Homework Week 5: 3 Ext Ext 3 ExtDocument10 pagesHomework Week 5: 3 Ext Ext 3 ExtIsabelle SimãoNo ratings yet
- TermodinâmicaDocument3 pagesTermodinâmicathiagopoetarnNo ratings yet
- Thermodynamic Process Equations and DiagramsDocument10 pagesThermodynamic Process Equations and DiagramsAbenayaNo ratings yet
- JEE Advanced Heat and Thermodynamics Important QuestionsDocument26 pagesJEE Advanced Heat and Thermodynamics Important QuestionsMayank VermaNo ratings yet
- Compressibility PDFDocument2 pagesCompressibility PDFJuan Daniel CabreraNo ratings yet
- ThermodynamicsDocument13 pagesThermodynamicsMonique OrugaNo ratings yet
- Thermodynamic Lesson 2Document7 pagesThermodynamic Lesson 2kelebekkNo ratings yet
- Formulas and Constants - Stp2022Document1 pageFormulas and Constants - Stp2022dayraNo ratings yet
- Problems303 1 SolDocument2 pagesProblems303 1 SolElaineNo ratings yet
- PVT Behavior of Fluida & EOSDocument53 pagesPVT Behavior of Fluida & EOSEka WahyuNo ratings yet
- Chapter8 Real Gases and Mixture of GasesDocument26 pagesChapter8 Real Gases and Mixture of GasesMUHAMMED FAISALNo ratings yet
- Termodinamikaa Maksimalni RadDocument30 pagesTermodinamikaa Maksimalni RadNataša AnđelićNo ratings yet
- Handouts PPE Day 3Document4 pagesHandouts PPE Day 3terrence miguel balitaNo ratings yet
- Physical proof of AM-GM inequality for 2 variables using thermodynamicsDocument2 pagesPhysical proof of AM-GM inequality for 2 variables using thermodynamicsVilakshan GuptaNo ratings yet
- Compressible Flow Through Nozzles and Diffusers: V DT V D V VDocument14 pagesCompressible Flow Through Nozzles and Diffusers: V DT V D V VCamilo SantacruzNo ratings yet
- Lecture 2 EDocument8 pagesLecture 2 EMihai MirceaNo ratings yet
- Meth HybridDocument80 pagesMeth HybridRuthneta JeffreyNo ratings yet
- Ideal GasDocument1 pageIdeal GasMike Raphy T. VerdonNo ratings yet
- Legea Lui Maxwell Pentru Distribuția Moleculelor După Vitezele Lor RelativeDocument3 pagesLegea Lui Maxwell Pentru Distribuția Moleculelor După Vitezele Lor RelativecristianNo ratings yet
- MEFT Plasma Physics Exercises CollectionDocument67 pagesMEFT Plasma Physics Exercises CollectionEsthefano Morales Campaña100% (1)
- ThermodynamicsDocument9 pagesThermodynamicssamir boseNo ratings yet
- CT 16Document39 pagesCT 16Bright ChabweraNo ratings yet
- Solution Part 3 (2023)Document9 pagesSolution Part 3 (2023)01khanh26No ratings yet
- Thermodynamics IDocument8 pagesThermodynamics IFerdaus Hasan BappiNo ratings yet
- 2PA35HCDocument3 pages2PA35HCMahdi GharibNo ratings yet
- Unit 2 First Law-Closed System ProblemsDocument11 pagesUnit 2 First Law-Closed System Problemspiravi66No ratings yet
- ET BSc02 22 09 20Document46 pagesET BSc02 22 09 20Roger MessiasNo ratings yet
- Termo 1' Modeli 8. BesmiriDocument11 pagesTermo 1' Modeli 8. BesmiriFeritNo ratings yet
- PHY303 Assignment 2 Solutions PDFDocument5 pagesPHY303 Assignment 2 Solutions PDFJohn McLovenNo ratings yet
- Carnot Engine Cycle AnalysisDocument6 pagesCarnot Engine Cycle AnalysisIuhence VergaraNo ratings yet
- Me2322 Thermo 20Document4 pagesMe2322 Thermo 20Zarina AdilbekovaNo ratings yet
- Thermodynamics All DerivationsDocument8 pagesThermodynamics All Derivationsanuragrabha99No ratings yet
- Fo 769 RmulasDocument1 pageFo 769 RmulasGabriela PierottiNo ratings yet
- Quiz 1 - MA SolutionsDocument6 pagesQuiz 1 - MA SolutionsabullaNo ratings yet
- Examples 1Document3 pagesExamples 1Prince MensahNo ratings yet
- Problems303 3 SolDocument2 pagesProblems303 3 SolliamfuentezNo ratings yet
- Thermodynamics Problems1-100Document5 pagesThermodynamics Problems1-100Monique OrugaNo ratings yet
- Thermo Chem Dynamics PawanDocument4 pagesThermo Chem Dynamics PawanPawan BabelNo ratings yet
- Solution Manual for an Introduction to Equilibrium ThermodynamicsFrom EverandSolution Manual for an Introduction to Equilibrium ThermodynamicsNo ratings yet
- Rishab IntroDocument4 pagesRishab IntroDeborsha DekaNo ratings yet
- XRF BR41146 Arl Optimx WDXRF SpectrometerDocument8 pagesXRF BR41146 Arl Optimx WDXRF SpectrometerGilson JoseNo ratings yet
- Lecture 10: Nucleic Acids (DNA & RNA)Document13 pagesLecture 10: Nucleic Acids (DNA & RNA)Binoni Laja EndongNo ratings yet
- X17Crni 16-2: C: 0,19 - 0,22 CR: 15,5 - 17,0 Ni: 1,5 - 2,5Document2 pagesX17Crni 16-2: C: 0,19 - 0,22 CR: 15,5 - 17,0 Ni: 1,5 - 2,5Aadhya engineering ServicesNo ratings yet
- Applications of Egg Shell and Egg Shell Membrane As AdsorbentsDocument13 pagesApplications of Egg Shell and Egg Shell Membrane As AdsorbentsAhmed AliNo ratings yet
- Sika®-1: Product Data SheetDocument2 pagesSika®-1: Product Data SheetKhin Sandi KoNo ratings yet
- Bulletin 114 IOM Manual GP Steam DsfilterDocument4 pagesBulletin 114 IOM Manual GP Steam DsfiltervextersNo ratings yet
- Pamphlet TG-X SeriesDocument2 pagesPamphlet TG-X SeriesrajeshNo ratings yet
- Bio Energiser Hair Boost Caffeine Power Tonic MSDS October 2016Document3 pagesBio Energiser Hair Boost Caffeine Power Tonic MSDS October 2016Alexander JefferyNo ratings yet
- Tic Reinforced AmcDocument27 pagesTic Reinforced AmcarlyNo ratings yet
- Periodic Table Display Poster A4Document1 pagePeriodic Table Display Poster A4Sarah Khan100% (1)
- SDS Sodium GluconateDocument4 pagesSDS Sodium GluconatemeNo ratings yet
- Ultimate Cheatcode CematconDocument82 pagesUltimate Cheatcode CematconJulian Deleon100% (1)
- GSI SLV SFI Whitepaper EN PDFDocument9 pagesGSI SLV SFI Whitepaper EN PDFjayahasanNo ratings yet
- Protocols Cleaning Disinfection SterilizationDocument23 pagesProtocols Cleaning Disinfection SterilizationGeneSegoviaNo ratings yet
- Reinforcing Low-Grade Glulam Beams with GFRP RodsDocument12 pagesReinforcing Low-Grade Glulam Beams with GFRP RodsCălin MartonNo ratings yet
- Acid and Base Number by Color-Indicator Titration: Standard Test Method ForDocument7 pagesAcid and Base Number by Color-Indicator Titration: Standard Test Method ForLuis EnriqueNo ratings yet
- 82 72Document8 pages82 72Chetan B ArkasaliNo ratings yet
- Heat, Temperature, and Heat Transfer: Cornell Doodle Notes FREE SAMPLERDocument13 pagesHeat, Temperature, and Heat Transfer: Cornell Doodle Notes FREE SAMPLERShraddha PatelNo ratings yet
- EcoFlame B-971 MSDSDocument8 pagesEcoFlame B-971 MSDSZirve PolimerNo ratings yet
- Glucose Solution ViscosityDocument13 pagesGlucose Solution ViscosityThomas Teh Qian Hua100% (1)
- RMS 6th 2019Document14 pagesRMS 6th 2019angelgupta2303No ratings yet
- CR8047 Sulzer CompaX Short Report1Document20 pagesCR8047 Sulzer CompaX Short Report1DucVikingNo ratings yet
- Material SpecificationDocument5 pagesMaterial SpecificationMuthu GaneshNo ratings yet
- Question Phy 101Document19 pagesQuestion Phy 101Mohammed kashimNo ratings yet
- Carbobond 3028 1Document1 pageCarbobond 3028 1Yan TengNo ratings yet
- Electronic Structure Calculations For Solids and Molecules: Theory and Computational MethodsDocument387 pagesElectronic Structure Calculations For Solids and Molecules: Theory and Computational MethodsJavier Gómez100% (1)
- Iriotec - 8850 - Merck - TDS (For Rest of The World) PDFDocument2 pagesIriotec - 8850 - Merck - TDS (For Rest of The World) PDFxy2zjgNo ratings yet
- SMAW LessonDocument8 pagesSMAW LessonOJ DogplaceNo ratings yet
- Pharmaceutical Gravimetric AnalysisDocument31 pagesPharmaceutical Gravimetric Analysiserorcrept100% (1)