You might also like
- BAB II NeurofibromaDocument14 pagesBAB II NeurofibromaIrfandy Chairi Sulaiman LubisNo ratings yet
- LP ObgynDocument13 pagesLP ObgynDoni Dhawa FaithfullNo ratings yet
- Insisional Biopsi Atas Indikasi Tumor MammaeDocument7 pagesInsisional Biopsi Atas Indikasi Tumor MammaeReni VildaNo ratings yet
- Teori WahamDocument17 pagesTeori WahamridhaniNo ratings yet
- Konsep Laporan Pendahuluan Gangren PedisDocument24 pagesKonsep Laporan Pendahuluan Gangren PedisAhmad ZainiNo ratings yet
- Tumor Jaringan LunakDocument11 pagesTumor Jaringan Lunakgiggs_libraNo ratings yet
- Kel 5 Kep. Gerontik - Sistem Indra (Katarak)Document38 pagesKel 5 Kep. Gerontik - Sistem Indra (Katarak)Anonymous 9auf5duNo ratings yet
- Pansitopenia dan EtiologinyaDocument22 pagesPansitopenia dan EtiologinyaChyntia UtamiNo ratings yet
- Alopecia AreataDocument28 pagesAlopecia AreataDefnityaVinorra100% (1)
- WOCDocument2 pagesWOCNadia Eka IndrianingNo ratings yet
- Makalah NevusDocument16 pagesMakalah NevusSinta RosNo ratings yet
- LP Diabetes MelitusDocument14 pagesLP Diabetes MelitusWina PradnyaNo ratings yet
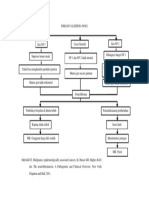
- Woc NeurofibromaDocument1 pageWoc Neurofibromaaris novenNo ratings yet
- HematogenInfeksiDocument4 pagesHematogenInfeksiNadia AfisaNo ratings yet
- Anatomi Fisiologi Dan Patofisiologi AbortusDocument8 pagesAnatomi Fisiologi Dan Patofisiologi AbortusTia AnggraeniNo ratings yet
- Laporan LBP R. Neuro (Isna Uwen)Document16 pagesLaporan LBP R. Neuro (Isna Uwen)Tuahenaidaaryani0% (1)
- Pathway CA ServiksDocument2 pagesPathway CA ServiksKurnilam Nur CiptaningsihNo ratings yet
- LP Fraktur Basis CraniiDocument33 pagesLP Fraktur Basis CraniiRuth Riani NainggolanNo ratings yet
- Satuan Acara Penyuluhan TetanusDocument6 pagesSatuan Acara Penyuluhan TetanusNovita SariNo ratings yet
- Askep Snake BiteDocument11 pagesAskep Snake BitePutra Endi PratamaNo ratings yet
- LAPORAN REFRAKSI MATADocument13 pagesLAPORAN REFRAKSI MATAKadek AstariniNo ratings yet
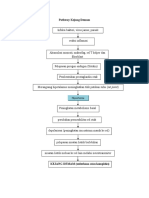
- Pathway Kejang Demam FixDocument2 pagesPathway Kejang Demam FixPutu.RirinNo ratings yet
- LP CvaDocument18 pagesLP CvaBiebaUmmuNo ratings yet
- LP Askep Mioma UteriDocument6 pagesLP Askep Mioma UteriSity MaryatulNo ratings yet
- BLEPHAROCHALASISDocument14 pagesBLEPHAROCHALASISAndi AlifyaNo ratings yet
- CEDEKRANDocument28 pagesCEDEKRANRobby Wiranata WijayaNo ratings yet
- Hipo Ke CKDDocument2 pagesHipo Ke CKDFitria Marina Sandy100% (1)
- LP AubDocument18 pagesLP Aubsheilapriyanti97No ratings yet
- Ikterus KholastatikDocument17 pagesIkterus KholastatikNur Syahraeny RamliNo ratings yet
- BPH Pro TurpDocument28 pagesBPH Pro TurpAl-amien Madridista Ibnu-jakfarNo ratings yet
- LP Hipoglikemia BaruDocument16 pagesLP Hipoglikemia BaruIndah SafitriNo ratings yet
- LP Chelapgia KMB 2Document10 pagesLP Chelapgia KMB 2idealti ajeengsNo ratings yet
- Penanggulangan Sakit Kepala VaskulerDocument38 pagesPenanggulangan Sakit Kepala VaskulerRAden Altaf Wibowo PutraNo ratings yet
- Edukasi Pencegahan Penyalahgunaan NAPZADocument16 pagesEdukasi Pencegahan Penyalahgunaan NAPZAMandira Musliana100% (1)
- Woc Tumor Otak PDFDocument2 pagesWoc Tumor Otak PDFMaria Stefani AsuatNo ratings yet
- Tumor PalpebraDocument58 pagesTumor Palpebrawadejack100% (1)
- Pathway AllDocument17 pagesPathway AllAyu FafaNo ratings yet
- LP - Anemia - Muliani Fix AccDocument32 pagesLP - Anemia - Muliani Fix AccMulianiNo ratings yet
- Penatalaksanaan Otitis EksternaDocument4 pagesPenatalaksanaan Otitis EksternaAlmira MeidaNo ratings yet
- Makalah HAPDocument20 pagesMakalah HAPNia Kurnia YuliantiNo ratings yet
- Makalah Blok 29 Syok HipovolemikDocument24 pagesMakalah Blok 29 Syok HipovolemikTiara Sari Irianti100% (1)
- TumorOtakDocument8 pagesTumorOtakDewi SartikaNo ratings yet
- Laporan Pendahuluan Rasa Aman Dan NyamanDocument13 pagesLaporan Pendahuluan Rasa Aman Dan NyamanLia SusandikaNo ratings yet
- GinjalDocument26 pagesGinjaleN Alimin100% (1)
- Mekanisme HipersensitivitasDocument4 pagesMekanisme HipersensitivitasNila Khurin'inNo ratings yet
- Meningitis LaporanDocument17 pagesMeningitis LaporanTuti NingsihNo ratings yet
- Sap EpilepsiDocument5 pagesSap EpilepsiAlifa RamadantiNo ratings yet
- Ringkas Parkinson 3A dan 3BDocument75 pagesRingkas Parkinson 3A dan 3BPreloved BycintaNo ratings yet
- LP Psoriasis NandaTiaraAgustin Tk2A D3KepDocument25 pagesLP Psoriasis NandaTiaraAgustin Tk2A D3KepNandaNo ratings yet
- Makalah Fraktur Femur DextraDocument15 pagesMakalah Fraktur Femur DextraMerissaArvianaNo ratings yet
- KarsinoidApendiksTumorJarangMetastasisDocument2 pagesKarsinoidApendiksTumorJarangMetastasisAchmad ShidiqNo ratings yet
- Laporan Pendahuluan Anemia AplastikDocument15 pagesLaporan Pendahuluan Anemia AplastikJy JhoeNo ratings yet
- Makalah Batu Empedu KholilDocument32 pagesMakalah Batu Empedu Kholilhei thereNo ratings yet
- Askep Neuroma Akustik 1Document8 pagesAskep Neuroma Akustik 1Stazyanda AlvozanNo ratings yet
- Miastenia GravisDocument33 pagesMiastenia GravisFernando HarisNo ratings yet
- Referat NeurofibromatosisDocument30 pagesReferat NeurofibromatosisAhmad Hashemi100% (2)
- Laporan Kasus Plexiform Neurofibroma RetroaurikularDocument32 pagesLaporan Kasus Plexiform Neurofibroma Retroaurikularchrystinayurita100% (1)
- LAPORAN KASUS NEUROFIBROMADocument16 pagesLAPORAN KASUS NEUROFIBROMABRI KUNo ratings yet
- NeurofibromaDocument28 pagesNeurofibromaTrias AdnyanaNo ratings yet
- Gangguan Penyakit Saraf TepiDocument19 pagesGangguan Penyakit Saraf TepiBustanoelNo ratings yet
- Surat Kuasa Tercucokmeong FixDocument1 pageSurat Kuasa Tercucokmeong FixRanda DpNo ratings yet
- Form Foto OmdDocument1 pageForm Foto OmdRanda DpNo ratings yet
- 13 Petunjuk Praktis Terapi Insulin Pada Pasien Diabetes MelitusDocument10 pages13 Petunjuk Praktis Terapi Insulin Pada Pasien Diabetes MelitusRanda DpNo ratings yet
- Petunjuk Praktis Terapi Insulin Pada Pasien Diabetes MelitusDocument41 pagesPetunjuk Praktis Terapi Insulin Pada Pasien Diabetes Melitusaidinasrul100% (28)
- Sri Apriyani (Thoraks PA)Document1 pageSri Apriyani (Thoraks PA)Randa DpNo ratings yet
- Soal Uas Anfis D4 2015Document5 pagesSoal Uas Anfis D4 2015Randa DpNo ratings yet
- Visi Misi Klinik Kadir MedikaDocument2 pagesVisi Misi Klinik Kadir MedikaRanda DpNo ratings yet
- Fix Mudah2anDocument1 pageFix Mudah2anRanda DpNo ratings yet
- 5148 - Daftar Peserta Internsip Rs Bhayangkara PalembangDocument3 pages5148 - Daftar Peserta Internsip Rs Bhayangkara PalembangRanda DpNo ratings yet
- Study Kasus KontrolDocument28 pagesStudy Kasus KontrolQONITA1722No ratings yet
- Surat Keterangan SakitDocument1 pageSurat Keterangan SakitRanda Dp0% (1)
- Mick Power (Thorax PA)Document1 pageMick Power (Thorax PA)Randa DpNo ratings yet
- Soal UasDocument1 pageSoal UasRanda DpNo ratings yet
- Surat Keterangan SakitDocument1 pageSurat Keterangan SakitRanda DpNo ratings yet
- Struktur Dan Fungsi SelDocument24 pagesStruktur Dan Fungsi SelRanda DpNo ratings yet
- Study Kasus KontrolDocument28 pagesStudy Kasus KontrolQONITA1722No ratings yet
- Hadi Wijaya (Thorax Pa)Document1 pageHadi Wijaya (Thorax Pa)Randa DpNo ratings yet
- Cardiomegali (LV La), Elongatio Aorta, Pulmo NDocument2 pagesCardiomegali (LV La), Elongatio Aorta, Pulmo NDessi AnugrahNo ratings yet
- Sudarto Bin Wudjatno (Thorax PA)Document1 pageSudarto Bin Wudjatno (Thorax PA)Randa DpNo ratings yet
- BAB II Perbedaan Posisi Tekanan DarahDocument62 pagesBAB II Perbedaan Posisi Tekanan DarahZul HermawanNo ratings yet
- AndrianyDocument1 pageAndrianyRanda DpNo ratings yet
- Fraktur ClaviculaDocument15 pagesFraktur ClaviculaAmanda PuspadewiNo ratings yet
- Contoh CT Scan KepalaDocument1 pageContoh CT Scan KepalaRanda DpNo ratings yet
- Zakaria Bin Komar (BNO)Document1 pageZakaria Bin Komar (BNO)Randa DpNo ratings yet
- Status Ujian Rehab - MedikDocument25 pagesStatus Ujian Rehab - MedikFebriWijayaNo ratings yet
- Surat Permohonan IjinDocument1 pageSurat Permohonan IjinRanda DpNo ratings yet
- PPT DSS BernadDocument17 pagesPPT DSS BernadRanda DpNo ratings yet
- BAB II Diare Ringan SedangDocument20 pagesBAB II Diare Ringan SedangRanda DpNo ratings yet
- Case GilutDocument21 pagesCase GilutRanda DpNo ratings yet
- BAB III SleDocument27 pagesBAB III SleRanda DpNo ratings yet