You might also like
- Ester Livshits and Roi Baer - A Well-Tempered Density Functional Theory of Electrons in MoleculesDocument10 pagesEster Livshits and Roi Baer - A Well-Tempered Density Functional Theory of Electrons in MoleculesNedsy8No ratings yet
- Away From Generalized Gradient Approximation: Orbital-Dependent Exchange-Correlation FunctionalsDocument18 pagesAway From Generalized Gradient Approximation: Orbital-Dependent Exchange-Correlation FunctionalsZohan Syah FatomiNo ratings yet
- Voskov2017 (OBL Isotermico. Add No Referencial Teorico)Document30 pagesVoskov2017 (OBL Isotermico. Add No Referencial Teorico)Igor LacerdaNo ratings yet
- Capecelatro Desjardins 2013 An Euler-Lagrange Strategy For Simulating Particle-Laden Flows PDFDocument31 pagesCapecelatro Desjardins 2013 An Euler-Lagrange Strategy For Simulating Particle-Laden Flows PDFAbgail PinheiroNo ratings yet
- Optimal Design of Barrel Vaults Using Charged Search SystemDocument8 pagesOptimal Design of Barrel Vaults Using Charged Search Systemgirish_deshmukh100% (1)
- Chebbi_16559Document28 pagesChebbi_16559acarrascotaberaNo ratings yet
- An Efficient and Near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster MethodDocument19 pagesAn Efficient and Near Linear Scaling Pair Natural Orbital Based Local Coupled Cluster MethodDuyen NguyenNo ratings yet
- The Sequential Optimal Attitude Recursion FilterDocument20 pagesThe Sequential Optimal Attitude Recursion FiltergantebrasNo ratings yet
- Journal of Electromagnetic Analysis and ApplicationsDocument7 pagesJournal of Electromagnetic Analysis and Applicationskrishnagaurav90No ratings yet
- Improved Particle Swarm Optimization Combined With Chaos: Bo Liu, Ling Wang, Yi-Hui Jin, Fang Tang, De-Xian HuangDocument11 pagesImproved Particle Swarm Optimization Combined With Chaos: Bo Liu, Ling Wang, Yi-Hui Jin, Fang Tang, De-Xian HuangmenguemengueNo ratings yet
- Becke, A.D., A New Mixing of Hartree-Fock and Local Density-Functional Theories, J. Chem. Phys. 96, 1372 (1993)Document7 pagesBecke, A.D., A New Mixing of Hartree-Fock and Local Density-Functional Theories, J. Chem. Phys. 96, 1372 (1993)RoyNo ratings yet
- Particle Swarm OptimizationDocument12 pagesParticle Swarm Optimizationjoseph676No ratings yet
- Modelos DFTDocument17 pagesModelos DFTAlberto Núñez CardezoNo ratings yet
- E195304 PDFDocument12 pagesE195304 PDFAzhar MahmoodNo ratings yet
- Density Functional Theory: An Introduction: Related ArticlesDocument12 pagesDensity Functional Theory: An Introduction: Related ArticlesAnangNo ratings yet
- Kaap ReprintDocument7 pagesKaap ReprintYoussef ToukaneNo ratings yet
- Geometry Optimization in Redundant Internal CoordinatesDocument6 pagesGeometry Optimization in Redundant Internal CoordinatesLorena Monterrosas PérezNo ratings yet
- Brief Study of XFEMDocument28 pagesBrief Study of XFEMVishal HotwaniNo ratings yet
- SCF Convergence and Chaos TheoryDocument4 pagesSCF Convergence and Chaos Theoryadu666No ratings yet
- Rappoport2010 - Property-Optimized Gaussian Basis Sets For Molecular ResponseDocument12 pagesRappoport2010 - Property-Optimized Gaussian Basis Sets For Molecular ResponseMateriais de química UFFNo ratings yet
- 2023-Physics Informed Neural Networks - A Case Study For Gas Transport ProblemsDocument16 pages2023-Physics Informed Neural Networks - A Case Study For Gas Transport ProblemsmaycvcNo ratings yet
- The Calculation of Ab Initio Molecular Geometries: Efficient Optimization by Natural Internal Coordinates and Empirical Correction by Offset ForcesDocument11 pagesThe Calculation of Ab Initio Molecular Geometries: Efficient Optimization by Natural Internal Coordinates and Empirical Correction by Offset ForcesLorena Monterrosas PérezNo ratings yet
- SSAdNDP Tool Deciphers Multi-Center Bonding in Periodic SystemsDocument8 pagesSSAdNDP Tool Deciphers Multi-Center Bonding in Periodic SystemsA Fernando CGNo ratings yet
- Guidance, Navigation & Control Conference ProceedingsDocument21 pagesGuidance, Navigation & Control Conference ProceedingsJORGE IVAN ZULUAGA CALLEJASNo ratings yet
- KrivovSV FreeEnergySurface RevProteinFolding MDS PLoS 2009 5 7 E1000428Document10 pagesKrivovSV FreeEnergySurface RevProteinFolding MDS PLoS 2009 5 7 E1000428jazmurdochNo ratings yet
- BioMolSim Chapter1 Johansson Et Al 2012-3-27Document26 pagesBioMolSim Chapter1 Johansson Et Al 2012-3-27nadeem akhtarNo ratings yet
- JChemPhys 117 7433Document16 pagesJChemPhys 117 7433g198817492No ratings yet
- J JMB 2004 06 058Document21 pagesJ JMB 2004 06 058Hamdan AhmadNo ratings yet
- Poulet 2003Document7 pagesPoulet 2003Fatin Farhan HaqueNo ratings yet
- Ab Initio Classical Trajectories On The Born-Oppenheimer Surface: Hessian - Based Integrators Using Fifth-Order Polynomial and Rational Function FitsDocument7 pagesAb Initio Classical Trajectories On The Born-Oppenheimer Surface: Hessian - Based Integrators Using Fifth-Order Polynomial and Rational Function FitsSeiyed Hamid Reza ShojaeiNo ratings yet
- 1 OnlineDocument14 pages1 OnlineGerardo HernandezNo ratings yet
- Paper 16Document18 pagesPaper 16Eng SaraaNo ratings yet
- Evolving Particle Swarm Optimization Implemented by A Genetic AlgorithmDocument2 pagesEvolving Particle Swarm Optimization Implemented by A Genetic AlgorithmAnup GaidhankarNo ratings yet
- Equation of State Calculations by Fast Computing Machines: Additional Information On J. Chem. PhysDocument7 pagesEquation of State Calculations by Fast Computing Machines: Additional Information On J. Chem. PhysIndra Prakash JhaNo ratings yet
- A Three-Dimensional, Longitudinally-Invariant Finite Element Model For Acoustic Propagation in Shallow Water WaveguidesDocument6 pagesA Three-Dimensional, Longitudinally-Invariant Finite Element Model For Acoustic Propagation in Shallow Water WaveguidesAsih RahmatNo ratings yet
- Revisiting The Finite Temperature String Method For The Calculation of Reaction Tubes and Free EnergiesDocument18 pagesRevisiting The Finite Temperature String Method For The Calculation of Reaction Tubes and Free Energiesakrito LeeNo ratings yet
- A Non-Standard Statistical Approach To The Silo Discharge: T E P JDocument8 pagesA Non-Standard Statistical Approach To The Silo Discharge: T E P JA94leo14hsetmaNo ratings yet
- Critical Examination of Incoherent Operations and A Physically Consistent Resource Theory of Quantum CoherenceDocument6 pagesCritical Examination of Incoherent Operations and A Physically Consistent Resource Theory of Quantum CoherenceAkshata MagarNo ratings yet
- Recursive Bayesian Estimation Using Gaussian SumsDocument15 pagesRecursive Bayesian Estimation Using Gaussian SumskhanziaNo ratings yet
- Shi & Eberhart 1998Document10 pagesShi & Eberhart 1998Dedy Junaidy Raya AluminiumNo ratings yet
- Examples of Colvars-based protocols: Association of polyleucine peptidesDocument5 pagesExamples of Colvars-based protocols: Association of polyleucine peptidesJorge Ramón Cantero PiñénezNo ratings yet
- Abinitio Tight BindingDocument25 pagesAbinitio Tight BindingRomario JulioNo ratings yet
- P.C. Bressloff and S. Coombes - Symmetry and Phase-Locking in A Ring of Pulse-Coupled Oscillators With Distributed DelaysDocument24 pagesP.C. Bressloff and S. Coombes - Symmetry and Phase-Locking in A Ring of Pulse-Coupled Oscillators With Distributed DelaysNeerFamNo ratings yet
- ADF-GLS Test - WikipediaDocument2 pagesADF-GLS Test - WikipediaVenu GopalNo ratings yet
- Density Functional TheoryDocument9 pagesDensity Functional TheoryVarovNo ratings yet
- Laurence E. Fried and Gregory S. Ezra - PERTURB: A Special-Purpose Algebraic Manipulation Program For Classical Perturbation TheoryDocument15 pagesLaurence E. Fried and Gregory S. Ezra - PERTURB: A Special-Purpose Algebraic Manipulation Program For Classical Perturbation TheoryOmasazzNo ratings yet
- PhysRevD 107 074509Document20 pagesPhysRevD 107 074509ghostNo ratings yet
- Tracking Multiple Optimizing SolutionsDocument16 pagesTracking Multiple Optimizing SolutionskarteekakNo ratings yet
- Computational Chemistry Using Modern Electronic Structure Method PDFDocument7 pagesComputational Chemistry Using Modern Electronic Structure Method PDFzan99No ratings yet
- Exchange Parameters From Approximate Self-Interaction Correction SchemeDocument10 pagesExchange Parameters From Approximate Self-Interaction Correction SchemeAkin AkandeNo ratings yet
- A Priori Phase Equilibrium Prediction From A Segment Contribution Solvation Model - Lin and SandlerDocument15 pagesA Priori Phase Equilibrium Prediction From A Segment Contribution Solvation Model - Lin and SandlerErick David Ravello SaldañaNo ratings yet
- Application of Implicit-Explicit High Order RungeDocument43 pagesApplication of Implicit-Explicit High Order RungeJosue AzurinNo ratings yet
- Simulation Study of Mass Transfer Coefficient in Slurry Bubble Column Reactor Using Neural NetworkDocument11 pagesSimulation Study of Mass Transfer Coefficient in Slurry Bubble Column Reactor Using Neural NetworkEmad ElsaidNo ratings yet
- Opf Ieee PDFDocument17 pagesOpf Ieee PDFMayank GoyalNo ratings yet
- Numerical Atomic Orbitals For Linear-Scaling Calculations: 共Received 6 April 2001; Published 28 November 2001兲Document9 pagesNumerical Atomic Orbitals For Linear-Scaling Calculations: 共Received 6 April 2001; Published 28 November 2001兲AytaçÇelikNo ratings yet
- Test-Particle Simulation of Space PlasmasDocument14 pagesTest-Particle Simulation of Space PlasmasSami UllahNo ratings yet
- Free Energy Calculations For Fluid and Solid Phases by - 2002 - Fluid Phase EquDocument8 pagesFree Energy Calculations For Fluid and Solid Phases by - 2002 - Fluid Phase EquAnonymous ypVNIINo ratings yet
- For Peer Review Only: The Coulson-Fischer Wave Function: Parametrization Using Distributed Gaussian Basis SetsDocument26 pagesFor Peer Review Only: The Coulson-Fischer Wave Function: Parametrization Using Distributed Gaussian Basis Setsבניה שטיינמץNo ratings yet
- A Birds-Eye View of Density Functional Theory - Klaus CapelleDocument69 pagesA Birds-Eye View of Density Functional Theory - Klaus CapelleIvanDeJ.OrnelasCruzNo ratings yet
- The Langevin and Generalised Langevin Approach to the Dynamics of Atomic, Polymeric and Colloidal SystemsFrom EverandThe Langevin and Generalised Langevin Approach to the Dynamics of Atomic, Polymeric and Colloidal SystemsNo ratings yet
- Electromagnetism: Physics 15bDocument9 pagesElectromagnetism: Physics 15bHome FisNo ratings yet
- Dipole Electric FieldDocument2 pagesDipole Electric FieldHome FisNo ratings yet
- PhysRevB 91 165404Document11 pagesPhysRevB 91 165404Home FisNo ratings yet
- Deformation and Scattering in Graphene Over Substrate Steps: 10.1103/physrevlett.108.096601Document4 pagesDeformation and Scattering in Graphene Over Substrate Steps: 10.1103/physrevlett.108.096601Home FisNo ratings yet
- Aeroporto Denver - MapaDocument1 pageAeroporto Denver - MapaHome FisNo ratings yet
- RevModPhys 86 153Document33 pagesRevModPhys 86 153Home FisNo ratings yet
- Topologial Phase TransitionsDocument12 pagesTopologial Phase TransitionsHome FisNo ratings yet
- Mimicking Nanoribbon Behavior With Graphene On A SiC SubstrateDocument4 pagesMimicking Nanoribbon Behavior With Graphene On A SiC SubstrateHome FisNo ratings yet
- Separation ProcessesDocument57 pagesSeparation ProcessesAngelo X0% (1)
- Remedial Cementing OverviewDocument31 pagesRemedial Cementing OverviewAbdul RehazkNo ratings yet
- GATE Previous Year Solved Papers Mechanical PDFDocument229 pagesGATE Previous Year Solved Papers Mechanical PDFRandy LeeNo ratings yet
- As Physics ISP MaterialsDocument33 pagesAs Physics ISP Materialsnazran68No ratings yet
- Polar Co-OrdinatesDocument10 pagesPolar Co-OrdinatesLourençoNo ratings yet
- S09 TTT DiagramDocument6 pagesS09 TTT Diagrampraba_343No ratings yet
- 1991 ServiceManual Mitsubishi 3000GT Vol 2Document316 pages1991 ServiceManual Mitsubishi 3000GT Vol 2NoAccount888100% (1)
- Tech Spot DC Vs CCDocument2 pagesTech Spot DC Vs CCLaura Bartlett100% (3)
- (2022) Enhancing Nonwoven Fabric Material Sound Absorption Using Embedded Labyrinthine Rigid StructuresDocument14 pages(2022) Enhancing Nonwoven Fabric Material Sound Absorption Using Embedded Labyrinthine Rigid StructuresIndra SiharNo ratings yet
- Intermolecular ForcesDocument23 pagesIntermolecular ForcesRonel LisingNo ratings yet
- Magnetrol Level Instruments - GWRDocument44 pagesMagnetrol Level Instruments - GWRVinodKumarNo ratings yet
- Huckel Molecular Orbital Theory: Sapan Kumar Jain Assistant Professor, JMIDocument45 pagesHuckel Molecular Orbital Theory: Sapan Kumar Jain Assistant Professor, JMIMayank GuptNo ratings yet
- Ke & Pe PracticeDocument2 pagesKe & Pe PracticeAlex DatsiukNo ratings yet
- Railway Track GeometryDocument88 pagesRailway Track Geometryarpit089No ratings yet
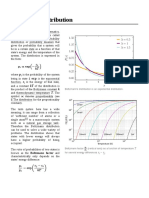
- Boltzmann DistributionDocument7 pagesBoltzmann Distributionhobson616No ratings yet
- LV Power MosfetDocument32 pagesLV Power MosfetAnonymous 2IdkjvoJNo ratings yet
- Magnetorheological Fluids As A Prospective Component of Composite ArmoursDocument5 pagesMagnetorheological Fluids As A Prospective Component of Composite ArmoursSuwerenNo ratings yet
- Leonard Susskind, Art Friedman - Special Relativity and Classical Field TheoDocument393 pagesLeonard Susskind, Art Friedman - Special Relativity and Classical Field TheoJuan Felipe Solorzano100% (7)
- 6 NSEP-TEST-6 Fluidsoundwave PCDocument11 pages6 NSEP-TEST-6 Fluidsoundwave PCShohom DeNo ratings yet
- Geo Metro DynamicsDocument23 pagesGeo Metro DynamicsRoogeliooNo ratings yet
- Wolfgang Pauli and The Fine-Structure ConstantDocument7 pagesWolfgang Pauli and The Fine-Structure ConstantMichael A. Sherbon100% (1)
- Day 2 PDFDocument64 pagesDay 2 PDFArabella SanchezNo ratings yet
- Effects of Lithium Carbonate Additives in High Alumina CastableDocument13 pagesEffects of Lithium Carbonate Additives in High Alumina CastableIan AjaNo ratings yet
- Mass Spectrometry Fundamental LC-MSDocument24 pagesMass Spectrometry Fundamental LC-MSdangerous067% (3)
- Forces and MotionDocument29 pagesForces and MotionMoeez Ahmad100% (1)
- Textbook On Engineering MechanicsDocument7 pagesTextbook On Engineering MechanicsPawan SinghNo ratings yet
- Monte Carlo Simulation of 1D Heisenberg ModelDocument12 pagesMonte Carlo Simulation of 1D Heisenberg Modelt_sairamNo ratings yet
- Cat Guia de Medicion de AislamientoDocument28 pagesCat Guia de Medicion de AislamientoMiguel SoteloNo ratings yet
- Tutorial Sheet 1 To 6Document11 pagesTutorial Sheet 1 To 6shubham sudarshanamNo ratings yet