You might also like
- Secme 23255Document127 pagesSecme 23255Thomas ParkerNo ratings yet
- Normas Coguanor Actualizado Dic2011Document75 pagesNormas Coguanor Actualizado Dic2011Paúl MonzónNo ratings yet
- Secme 23255Document127 pagesSecme 23255Thomas ParkerNo ratings yet
- Estudio de Tiempos para Linea Corrugado PDFDocument11 pagesEstudio de Tiempos para Linea Corrugado PDFThomas ParkerNo ratings yet
- Proyecto FinalDocument78 pagesProyecto FinalThomas ParkerNo ratings yet
- Productos LacteosDocument36 pagesProductos LacteosRoger Vera SandovalNo ratings yet
- Listado Servicios LaboratorioDocument7 pagesListado Servicios LaboratorioThomas ParkerNo ratings yet
- Ensayo de Elongacion y SelloDocument26 pagesEnsayo de Elongacion y SelloThomas Parker100% (1)
- FlexografiaDocument14 pagesFlexografiaThomas ParkerNo ratings yet
- FlexografiaDocument7 pagesFlexografiajuan100% (2)
- Industria FlexograficaDocument95 pagesIndustria FlexograficaJuan Pablo Azcuña C.No ratings yet
- Empaque coextruido: sello hermético para ventanas de aluminioDocument2 pagesEmpaque coextruido: sello hermético para ventanas de aluminioThomas ParkerNo ratings yet
- Implantación Del Sistema Smed en Un Proceso de Impresión FlexográficaDocument19 pagesImplantación Del Sistema Smed en Un Proceso de Impresión Flexográficaмiĸe мeиdozaNo ratings yet
- Manual de Normas de Calidad Envase y EmpaqueDocument91 pagesManual de Normas de Calidad Envase y EmpaqueAin't Almight100% (1)
- Manual para PreprensaDocument81 pagesManual para PreprensaJUANDAVID0313No ratings yet
- Fidel PDFDocument13 pagesFidel PDFRodrigo Roko MartinezNo ratings yet
- Boletin - Carnicos de EMPAQUESDocument76 pagesBoletin - Carnicos de EMPAQUESHeylinFernandézGutierrézNo ratings yet
- Manual de Normas de Calidad Envase y EmpaqueDocument91 pagesManual de Normas de Calidad Envase y EmpaqueAin't Almight100% (1)
- Libro Emparejarse Versión Digital PDFDocument124 pagesLibro Emparejarse Versión Digital PDFMarco Carranza33% (3)
- Gestion de RiesgosDocument17 pagesGestion de Riesgossharon gradosNo ratings yet
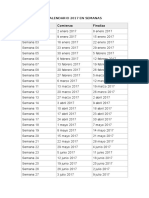
- Calendario SemanasDocument2 pagesCalendario SemanasThomas ParkerNo ratings yet
- Acta Constitucion LtdaDocument5 pagesActa Constitucion LtdaThomas ParkerNo ratings yet
- Indicadores de GestionDocument44 pagesIndicadores de GestionjuartoIINo ratings yet
- Vicente Lorenzo EstebanDocument190 pagesVicente Lorenzo EstebanThomas ParkerNo ratings yet
- Calidad TotalDocument52 pagesCalidad TotalRafael EduardoNo ratings yet
- Como Gerenciar PDFDocument12 pagesComo Gerenciar PDFIvan QuispeNo ratings yet
- Tesis - Isai Estudio de TiemposDocument157 pagesTesis - Isai Estudio de TiemposAlan Isai Valdez Castrejón100% (1)
- Polimeros 1.unlockedDocument54 pagesPolimeros 1.unlockedSergio GuillenNo ratings yet
- Dsesarrollo Del ProductoDocument4 pagesDsesarrollo Del ProductosandinochapinNo ratings yet
- MerckDocument11 pagesMerckJOSE CACERES FLOREZNo ratings yet
- FDA regula medicamentos botánicosDocument8 pagesFDA regula medicamentos botánicosmarcela floresNo ratings yet
- Por Que Comes Lo Que Comes Reflexiones Sobre La Alimentacion ModernaDocument7 pagesPor Que Comes Lo Que Comes Reflexiones Sobre La Alimentacion Modernaclauerimh7100% (1)
- Presentacion Minsal Administración de MedicamentosDocument3 pagesPresentacion Minsal Administración de MedicamentosCatalina Alejandra Pinto ArancibiaNo ratings yet
- Prótesis caninaDocument28 pagesPrótesis caninaluis p guerreroNo ratings yet
- Entregable 12Document8 pagesEntregable 12JHON ANTHONY CCOECCA REBOLLARNo ratings yet
- Lo Que Necesitas Saber Del FSMA - AIBDocument20 pagesLo Que Necesitas Saber Del FSMA - AIBNatali Pérez AguirreNo ratings yet
- Requisitos Clave FsmaDocument3 pagesRequisitos Clave FsmaMayra CortesNo ratings yet
- 3er-Avance-Mili, Lisbeth y FlorDocument67 pages3er-Avance-Mili, Lisbeth y FlorKevin Aldair Pacheco HuamanNo ratings yet
- Preguntas FrecuentesDocument15 pagesPreguntas FrecuentesYennifer Castillo CastanoNo ratings yet
- Trabajo de Campo MarketimgDocument35 pagesTrabajo de Campo MarketimgBrenda Martinez AtaoNo ratings yet
- Alerta 76-22Document2 pagesAlerta 76-22Q.F. Santiago AlarcónNo ratings yet
- Tarea Ejercicios Anderson PATRICIA GONZALEZDocument15 pagesTarea Ejercicios Anderson PATRICIA GONZALEZpatricia100% (1)
- Documento Unico para EstudioDocument4 pagesDocumento Unico para EstudioCarolina ApazaNo ratings yet
- Carne de Conejo PDFDocument3 pagesCarne de Conejo PDFMariano Spena ChefNo ratings yet
- Ahumado (Es)Document28 pagesAhumado (Es)Beker Malpartida TolentinoNo ratings yet
- BIOEQUIVALENCIA. PRACTICA 3 Final A PDFDocument14 pagesBIOEQUIVALENCIA. PRACTICA 3 Final A PDFRobocopNo ratings yet
- Alimentos Funcionales - International Food Information CouncilDocument1 pageAlimentos Funcionales - International Food Information CouncilTCNo ratings yet
- La plata coloidal ayuda a la salud de los gatosDocument16 pagesLa plata coloidal ayuda a la salud de los gatosEpiphania de MoiNo ratings yet
- 1 Laura Pasculli INVIMADocument24 pages1 Laura Pasculli INVIMAcamadire1584No ratings yet
- Guia Controles Preventivos APEAM 1Document69 pagesGuia Controles Preventivos APEAM 1Yeny Ceballos100% (1)
- Wafae Rezquellah - THESIS Validacion de LimpiezaDocument237 pagesWafae Rezquellah - THESIS Validacion de Limpiezaezygadlo6492No ratings yet
- Healing Is Voltage - The Handbook (PDFDrive) 1Document443 pagesHealing Is Voltage - The Handbook (PDFDrive) 1JORGE100% (6)
- Exportación de semillas de sésamo a EEUU: Análisis de mercadoDocument20 pagesExportación de semillas de sésamo a EEUU: Análisis de mercadoJuany Castillo VeronNo ratings yet
- Consideraciones para realizar pruebas exentasDocument60 pagesConsideraciones para realizar pruebas exentasAngelica UribeNo ratings yet
- 50 Razones para Oponerse A La Fluoración Del Agua PotableDocument17 pages50 Razones para Oponerse A La Fluoración Del Agua Potablecme1020No ratings yet
- Fda Food Code en EspañolDocument868 pagesFda Food Code en EspañolSophie Tueros Giler100% (2)
- Reglamento técnico aditivos alimentos ColombiaDocument6 pagesReglamento técnico aditivos alimentos ColombiaFlor Marina Achury DelgadoNo ratings yet
- Documental The Bleeding Edge Diego MartínezDocument7 pagesDocumental The Bleeding Edge Diego MartínezDiego MartínezNo ratings yet
- Trabajo Miel de Abeja peru-EE - UUDocument15 pagesTrabajo Miel de Abeja peru-EE - UUcriztian lopezNo ratings yet