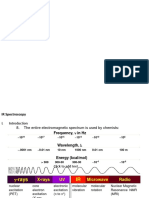
You might also like
- Detect molecular functionality with IR spectroscopyDocument41 pagesDetect molecular functionality with IR spectroscopyDiah Ayu KinasihNo ratings yet
- IR SPECTROSCOPY OrganicDocument46 pagesIR SPECTROSCOPY OrganicSumit VermaNo ratings yet
- 7 - Vibrational - Spectro - IR Spectroscopy - Adapted - Fall2021 - v1Document56 pages7 - Vibrational - Spectro - IR Spectroscopy - Adapted - Fall2021 - v1arwaNo ratings yet
- Infrared Spectroscopy: Nur Jannah BaturanteDocument17 pagesInfrared Spectroscopy: Nur Jannah BaturanteFatmaWati AlfikarNo ratings yet
- UntitledDocument88 pagesUntitledPutri Diana BintangNo ratings yet
- Infrared Spectroscopy: Nyi Mekar SaptariniDocument56 pagesInfrared Spectroscopy: Nyi Mekar SaptariniKita RadisaNo ratings yet
- KA 2023 IR KeduaDocument37 pagesKA 2023 IR KeduaTrianita SilaenNo ratings yet
- Advanced PH Analysis Lec 3&4Document60 pagesAdvanced PH Analysis Lec 3&4knowlegebook6No ratings yet
- Advanced PH Analysis Lec3 IR SpectrosDocument26 pagesAdvanced PH Analysis Lec3 IR Spectrosknowlegebook6No ratings yet
- CHMBD 449 - Organic Spectral: AnalysisDocument18 pagesCHMBD 449 - Organic Spectral: AnalysisIleana ManciuleaNo ratings yet
- Infrared SpectrosDocument25 pagesInfrared SpectrosSnehal MohireNo ratings yet
- Spectroscopy - B.Tech First YearDocument26 pagesSpectroscopy - B.Tech First YeartifinNo ratings yet
- IR SpectrosDocument39 pagesIR SpectrosYurîiGislãineNo ratings yet
- Infrared SpectrosDocument50 pagesInfrared SpectrosmohammedabubakrNo ratings yet
- IR SpectrosDocument119 pagesIR SpectrosRojan PradhanNo ratings yet
- Infrared Spectroscopy - Ch2Document71 pagesInfrared Spectroscopy - Ch2Ayat SbeihNo ratings yet
- General Introduction Molecular Spectroscopy BSC Lect1 2017Document34 pagesGeneral Introduction Molecular Spectroscopy BSC Lect1 2017Èmâan MahHeërrNo ratings yet
- IR SpectrosDocument96 pagesIR SpectrosMOHAMMED ABDUL HAINo ratings yet
- UV - Visible SpectrophotometryDocument171 pagesUV - Visible SpectrophotometryGudisa KufoNo ratings yet
- Vibrational (IR) SpectrosDocument28 pagesVibrational (IR) SpectrosSajith KurianNo ratings yet
- Chm557 Chapter 3Document70 pagesChm557 Chapter 3Nurul izzatiNo ratings yet
- SpectrosDocument47 pagesSpectrosAkhilesh PandeyNo ratings yet
- New Microsoft Office Word DocumentDocument39 pagesNew Microsoft Office Word DocumentUadNo ratings yet
- Pengantar ANSPEKDocument56 pagesPengantar ANSPEKFlorencia AngellicaNo ratings yet
- Beginning of Quantum AgeDocument7 pagesBeginning of Quantum Agespidy warriorsNo ratings yet
- Lct-1 General Introduction - 2019Document38 pagesLct-1 General Introduction - 2019Abdelfattah Mohamed OufNo ratings yet
- Notes 14C IRDocument6 pagesNotes 14C IRLouie SyNo ratings yet
- IR NovDocument53 pagesIR Novmsk3kiidNo ratings yet
- Analytical LastDocument309 pagesAnalytical Lastshamsu mahammadNo ratings yet
- Chem 442 - Chapter OneDocument26 pagesChem 442 - Chapter OneArindam DasNo ratings yet
- FTIR Lecture SlidesDocument25 pagesFTIR Lecture Slidesabdul rehman khanNo ratings yet
- CHM556/ CHM557 Organic Chemistry Ii/ Organic Chemistry: Spectroscopy of Carbon CompoundsDocument200 pagesCHM556/ CHM557 Organic Chemistry Ii/ Organic Chemistry: Spectroscopy of Carbon CompoundsnanaNo ratings yet
- Electronic Radiation SpectrumDocument36 pagesElectronic Radiation SpectrumhananNo ratings yet
- Spectroscopy Unit 1Document19 pagesSpectroscopy Unit 1veluselvamaniNo ratings yet
- Reading: Vibrational Spectroscopy Revised: 2/24/15Document9 pagesReading: Vibrational Spectroscopy Revised: 2/24/15abdooufNo ratings yet
- 1.1 AH CfE Chemistry NotesDocument7 pages1.1 AH CfE Chemistry Noteskira.zavyalova24No ratings yet
- Physics: Autonomous Educational Organization Nazarbayev Intellectual SchoolsDocument49 pagesPhysics: Autonomous Educational Organization Nazarbayev Intellectual SchoolsAbylai Tashtan100% (1)
- IR Specroscopy - 2Document16 pagesIR Specroscopy - 2Fatima AhmedNo ratings yet
- SpectrosDocument99 pagesSpectrosBikramNo ratings yet
- CY1004 - Spectroscopy - Till RotationDocument46 pagesCY1004 - Spectroscopy - Till Rotationasr8948222209No ratings yet
- Chapter 1 Full Chm556 2018Document206 pagesChapter 1 Full Chm556 2018Sabrina100% (1)
- Introduction to SpectroscopyDocument5 pagesIntroduction to Spectroscopybinfa kashafNo ratings yet
- Unit 1 Electromagnetic Radiation-1Document23 pagesUnit 1 Electromagnetic Radiation-1Saif YounusNo ratings yet
- 8-X RaysDocument6 pages8-X Raysnitdhakar04No ratings yet
- Instrumentation Part 1Document5 pagesInstrumentation Part 1Johanna Rose Cobacha SalvediaNo ratings yet
- PHYSICS Grade 12Document48 pagesPHYSICS Grade 12Андрей ТимонинNo ratings yet
- Infrared Spectroscopy Reveals Functional GroupsDocument16 pagesInfrared Spectroscopy Reveals Functional GroupsFatima AhmedNo ratings yet
- eBook+Introduction+to+Spectroscopy Pavis 32 121Document7 pageseBook+Introduction+to+Spectroscopy Pavis 32 121devi dwi rustaminingrumNo ratings yet
- Theoryofirspectroscopy 160622080111Document50 pagesTheoryofirspectroscopy 160622080111ArshNo ratings yet
- Light and Illumination: A PowerPoint Presentation on Fundamental ConceptsDocument31 pagesLight and Illumination: A PowerPoint Presentation on Fundamental ConceptsVasavi VaasuNo ratings yet
- Obt751 - Analytical Methods and Instrumentation Lecture - 2Document28 pagesObt751 - Analytical Methods and Instrumentation Lecture - 2Jayashree Sathiyanarayanan100% (1)
- Chap 3 Spectroscopy 1Document93 pagesChap 3 Spectroscopy 1Irfan AzaharNo ratings yet
- L3-4. UV SpectrosDocument33 pagesL3-4. UV Spectrosngocnm.bi12-320No ratings yet
- Infrared and UVVis SpectrosDocument46 pagesInfrared and UVVis SpectrosOlivia ChoiNo ratings yet
- Unit4, Atomic&MolecularPhysics, InfraRed Spectros PDFDocument43 pagesUnit4, Atomic&MolecularPhysics, InfraRed Spectros PDFanujjuetNo ratings yet
- Spectrophotometric Principle and ApplicationsDocument22 pagesSpectrophotometric Principle and ApplicationsIfty KharulNo ratings yet
- Intensity of Electromagnetic Waves as a Function of Frequency, Source Distance and Aperture AngleFrom EverandIntensity of Electromagnetic Waves as a Function of Frequency, Source Distance and Aperture AngleNo ratings yet
- Solid-State Circuits: Electrical Engineering DivisonFrom EverandSolid-State Circuits: Electrical Engineering DivisonRating: 4.5 out of 5 stars4.5/5 (4)
- Challenges To Healthy Food Choices Can Include Food Insecurity Which Is AnDocument2 pagesChallenges To Healthy Food Choices Can Include Food Insecurity Which Is AnRohit SinghNo ratings yet
- B18103 Rohit Singh CVDocument1 pageB18103 Rohit Singh CVRohit SinghNo ratings yet
- Challenges To Healthy Food Choices Can Include Food Insecurity Which Is AnDocument1 pageChallenges To Healthy Food Choices Can Include Food Insecurity Which Is AnRohit SinghNo ratings yet
- MNLDocument86 pagesMNLRohit SinghNo ratings yet
- Events Certi 13Document1 pageEvents Certi 13Rohit SinghNo ratings yet
- Optical DistorttionDocument2 pagesOptical DistorttionRohit SinghNo ratings yet
- Operations Management EssentialsDocument24 pagesOperations Management EssentialsRohit SinghNo ratings yet
- Course Outline BIDMDocument7 pagesCourse Outline BIDMRohit SinghNo ratings yet
- Principles of Corporate Finance: 6th EditionDocument4 pagesPrinciples of Corporate Finance: 6th EditionRohit SinghNo ratings yet
- Challenges To Healthy Food Choices Can Include Food Insecurity Which Is AnDocument1 pageChallenges To Healthy Food Choices Can Include Food Insecurity Which Is AnRohit SinghNo ratings yet
- Untitled 2Document2 pagesUntitled 2Rohit SinghNo ratings yet
- Organic Compound Identification Using Infrared SpectrosDocument34 pagesOrganic Compound Identification Using Infrared SpectrosRohit SinghNo ratings yet
- CHPT 04Document5 pagesCHPT 04Rohit SinghNo ratings yet
- Two SampleDocument16 pagesTwo SampleRohit SinghNo ratings yet
- Sampling RandomDocument11 pagesSampling RandomRohit SinghNo ratings yet
- Two SampleDocument2 pagesTwo SampleRohit SinghNo ratings yet
- Dont Lose FocusDocument9 pagesDont Lose FocusRohit Singh100% (1)
- DakotaDocument4 pagesDakotaRohit SinghNo ratings yet
- Oct 17, 2018 Plan For The Next 4.5 Days Use These WiselyDocument1 pageOct 17, 2018 Plan For The Next 4.5 Days Use These WiselyRohit SinghNo ratings yet
- SamplingDocument148 pagesSamplingRohit SinghNo ratings yet
- Confidence IntervalDocument2 pagesConfidence IntervalRohit SinghNo ratings yet
- END TERM Answers Prev YearDocument20 pagesEND TERM Answers Prev YearRohit SinghNo ratings yet
- Batt Et Al Inclined Sedimentati PDFDocument7 pagesBatt Et Al Inclined Sedimentati PDFRohit SinghNo ratings yet
- Confidence IntervalDocument2 pagesConfidence IntervalRohit SinghNo ratings yet
- SamplingDocument148 pagesSamplingRohit SinghNo ratings yet
- Syllabus For The End Term ExamDocument1 pageSyllabus For The End Term ExamRohit SinghNo ratings yet
- SamplingDocument148 pagesSamplingRohit SinghNo ratings yet
- CaseDocument1 pageCaseRohit SinghNo ratings yet
- Aug 14Document2 pagesAug 14Rohit SinghNo ratings yet
- Electrolysis Class XIDocument17 pagesElectrolysis Class XIPrashantNo ratings yet
- Spe 115 G PDFDocument14 pagesSpe 115 G PDFJuan SantosNo ratings yet
- Chemical Equilibrium MCQDocument13 pagesChemical Equilibrium MCQNidhi SisodiaNo ratings yet
- 3M Fluorinert Liquids For Electronics ManufacturingDocument4 pages3M Fluorinert Liquids For Electronics ManufacturingIon ZabetNo ratings yet
- Chp07ans PDFDocument28 pagesChp07ans PDFBinit KarNo ratings yet
- Nernst EquationDocument10 pagesNernst Equationaceyourchemistry.blogspot.com.sgNo ratings yet
- Magnetic RefrigerationDocument21 pagesMagnetic RefrigerationVishnu RajuNo ratings yet
- Pyrolysis of Plastic WasteDocument12 pagesPyrolysis of Plastic Wasteup4all100% (4)
- Magnetic Refrigeration SeminarDocument29 pagesMagnetic Refrigeration SeminarIdulla Birajdar100% (2)
- Chemistry Project: Done by - Viral, Manan, Kush, Naitik, Parthiv, VirajDocument12 pagesChemistry Project: Done by - Viral, Manan, Kush, Naitik, Parthiv, VirajViraj PatelNo ratings yet
- Inquiry LabDocument2 pagesInquiry Labapi-568508101No ratings yet
- Hydraulic FluidsDocument20 pagesHydraulic FluidsRamirez Indeleble100% (1)
- Determination of Calofric Value of The BiodieselDocument24 pagesDetermination of Calofric Value of The BiodieselYaman DurmuşNo ratings yet
- AlcoholDocument45 pagesAlcoholMichaelWongNo ratings yet
- ConductivitySensor BrochureDocument4 pagesConductivitySensor BrochureJames TakeNo ratings yet
- Electron Configuration 2Document6 pagesElectron Configuration 2268953No ratings yet
- Introduction to Compressible Gas Flow DynamicsDocument2 pagesIntroduction to Compressible Gas Flow DynamicsMeetu KaurNo ratings yet
- Glenium® Ace 30: Description and Field of Application Zero Energy SystemDocument2 pagesGlenium® Ace 30: Description and Field of Application Zero Energy SystemFrancois-No ratings yet
- Mr. Manish Kharwade - PrajDocument41 pagesMr. Manish Kharwade - PrajNithi AnandNo ratings yet
- Pr1 - Dr. Siti Nurul 'Ain Yusop - Norhafizah Binti Ismail NasiruddinDocument4 pagesPr1 - Dr. Siti Nurul 'Ain Yusop - Norhafizah Binti Ismail NasiruddinNafiz SyadhamierNo ratings yet
- Faraday's Law WorksheetDocument4 pagesFaraday's Law WorksheetBrianna MalcolmNo ratings yet
- Gas Turbine GEK28143aDocument10 pagesGas Turbine GEK28143aDede Maulana100% (1)
- Lecture Notes Enzyme 2 Enzyme Kinetics WebDocument29 pagesLecture Notes Enzyme 2 Enzyme Kinetics WebAldren RebaLdeNo ratings yet
- Methods of Sulphuric AcidDocument18 pagesMethods of Sulphuric AcidKrushit PatelNo ratings yet
- PARTICLE PROPERTIES OF WAVESDocument27 pagesPARTICLE PROPERTIES OF WAVESDikshit AjitsariaNo ratings yet
- Answers To Science Focus 3 Coursebook QuestionsDocument61 pagesAnswers To Science Focus 3 Coursebook QuestionsrobouNo ratings yet
- Ourse: CHE442 - SEPARATION PROCESS 1 (3 Credits /compulsory)Document8 pagesOurse: CHE442 - SEPARATION PROCESS 1 (3 Credits /compulsory)Anonymous TUXPKUSRDUNo ratings yet
- F3654870GATE-Chemical Engineering Previous Paper 2001Document10 pagesF3654870GATE-Chemical Engineering Previous Paper 2001Shashank bhattNo ratings yet
- Chapter - 3.1 - Finale External Forced ConvectionDocument18 pagesChapter - 3.1 - Finale External Forced ConvectioneirinaNo ratings yet
- Chemical Bonding ExplainedDocument10 pagesChemical Bonding ExplainedMYLENE B. ZABALLERONo ratings yet