You might also like
- 2009 Brief Bioinform 10 378-391Document14 pages2009 Brief Bioinform 10 378-391mbrylinskiNo ratings yet
- 2011 J Struct Biol 173 558-569Document12 pages2011 J Struct Biol 173 558-569mbrylinskiNo ratings yet
- 2008 Proteins 70 363-377Document15 pages2008 Proteins 70 363-377mbrylinskiNo ratings yet
- Struct GenomDocument7 pagesStruct GenomNaidelyn HernandezNo ratings yet
- Brylinski. FindsiteDocument6 pagesBrylinski. FindsiteRina HerowatiNo ratings yet
- Zhou 105Document11 pagesZhou 105suryasanNo ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- 2013 Front Genet 4 118Document7 pages2013 Front Genet 4 118mbrylinskiNo ratings yet
- A Comprehensive Analysis of The Structure-Function Relationship in Proteins Based On Local Structure SimilarityDocument9 pagesA Comprehensive Analysis of The Structure-Function Relationship in Proteins Based On Local Structure SimilarityselvaparuNo ratings yet
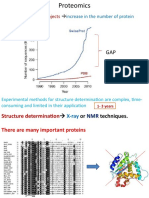
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- s12859-021-04323Document20 pagess12859-021-04323Me KhNo ratings yet
- Protein Loop Closure Using Orientational Restraints from NMR DataDocument21 pagesProtein Loop Closure Using Orientational Restraints from NMR Datakbarn389No ratings yet
- Function Prediction Using Neighborhood Patterns: Petko Bogdanov Petko@cs - Ucsb.edu Ambuj Singh Ambuj@cs - Ucsb.eduDocument7 pagesFunction Prediction Using Neighborhood Patterns: Petko Bogdanov Petko@cs - Ucsb.edu Ambuj Singh Ambuj@cs - Ucsb.eduSovanSahaNo ratings yet
- Protein Bioinformatics: From Sequence to FunctionFrom EverandProtein Bioinformatics: From Sequence to FunctionRating: 4.5 out of 5 stars4.5/5 (2)
- Paper1 Decodingdlforbinfdingaffinit1yDocument32 pagesPaper1 Decodingdlforbinfdingaffinit1yGANYA U 2022 Batch,PES UniversityNo ratings yet
- Assessment of Blind Predictions of Protein-Protein Interactions: Current Status of Docking MethodsDocument17 pagesAssessment of Blind Predictions of Protein-Protein Interactions: Current Status of Docking Methodsravi_guluniNo ratings yet
- Efficient Protein Interaction Hotspot Identification in ProteinsDocument4 pagesEfficient Protein Interaction Hotspot Identification in ProteinsIJERDNo ratings yet
- COACH YZhangDocument8 pagesCOACH YZhangChristos FeidakisNo ratings yet
- Cavidad PrediccionDocument9 pagesCavidad PrediccionLuis MartinezNo ratings yet
- 2014 J Mol Recognit 28 35-48Document14 pages2014 J Mol Recognit 28 35-48mbrylinskiNo ratings yet
- Protein Interaction Mol DockingDocument49 pagesProtein Interaction Mol DockingKhuwaylaNo ratings yet
- Mustang PSFB FinalDocument49 pagesMustang PSFB FinalNiranjan BhuvanaratnamNo ratings yet
- 2010 Bioinformatics 26 687-688Document2 pages2010 Bioinformatics 26 687-688mbrylinskiNo ratings yet
- Cyclic peptide structure prediction and design using AlphaFoldDocument25 pagesCyclic peptide structure prediction and design using AlphaFoldxinyuwu0528No ratings yet
- Statistics for Bioinformatics: Methods for Multiple Sequence AlignmentFrom EverandStatistics for Bioinformatics: Methods for Multiple Sequence AlignmentNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- Position-Specific Scoring Matrix for Protein Function PredictionDocument8 pagesPosition-Specific Scoring Matrix for Protein Function PredictionW Antonio Muñoz ChNo ratings yet
- Proteins - 2007 - Mintseris - Integrating Statistical Pair Potentials Into Protein Complex PredictionDocument10 pagesProteins - 2007 - Mintseris - Integrating Statistical Pair Potentials Into Protein Complex PredictionInes MejriNo ratings yet
- Leis 2010Document13 pagesLeis 2010iqbal iqbalNo ratings yet
- Protein Unfolding Behavior Studied by Elastic Network ModelDocument11 pagesProtein Unfolding Behavior Studied by Elastic Network ModeljmxnetdjNo ratings yet
- Protein Structure Prediction TechniquesDocument30 pagesProtein Structure Prediction TechniquesJhanvi SNo ratings yet
- 2015 Article 14 Twilight ZoneDocument11 pages2015 Article 14 Twilight Zoneadiav PakNo ratings yet
- Boolean gene regulatory network model of centromere functionDocument17 pagesBoolean gene regulatory network model of centromere functionUriHaimNo ratings yet
- Algorithms and Methods in Structural Bioinformatics (Computational Biology) (Nurit Haspel (Editor) Etc.)Document120 pagesAlgorithms and Methods in Structural Bioinformatics (Computational Biology) (Nurit Haspel (Editor) Etc.)Sathish KumarNo ratings yet
- SESNetDocument19 pagesSESNetThales WillianNo ratings yet
- TMP D73Document15 pagesTMP D73FrontiersNo ratings yet
- Nucl. Acids Res. 2005 Ginalski 1874 91Document18 pagesNucl. Acids Res. 2005 Ginalski 1874 91Rizki PerdanaNo ratings yet
- Models and Formalisms For Systems BiologyDocument5 pagesModels and Formalisms For Systems BiologydrsharghiNo ratings yet
- Protein Structure Prediction ThesisDocument8 pagesProtein Structure Prediction ThesisWriteMyThesisPaperManchester100% (2)
- Mol - Modelling of BCL-2 FinalDocument37 pagesMol - Modelling of BCL-2 FinalVannakumar SubramanianNo ratings yet
- Homo LogyDocument8 pagesHomo LogyIgnacioEguinoaNo ratings yet
- New Approaches of Protein Function Prediction from Protein Interaction NetworksFrom EverandNew Approaches of Protein Function Prediction from Protein Interaction NetworksNo ratings yet
- HardwareDocument16 pagesHardwarepaulogameiroNo ratings yet
- Poing PDFDocument16 pagesPoing PDFAanNo ratings yet
- Unit 1: Structural GenomicsDocument4 pagesUnit 1: Structural GenomicsLavanya ReddyNo ratings yet
- (Eisenberg@@) (2000) (NA) (X) Protein Function PDFDocument4 pages(Eisenberg@@) (2000) (NA) (X) Protein Function PDFbyominNo ratings yet
- Logical Modeling of Biological SystemsFrom EverandLogical Modeling of Biological SystemsLuis Fariñas del CerroNo ratings yet
- Protein Sequence Similarity Analysis Using Frequency Domain ApproachDocument14 pagesProtein Sequence Similarity Analysis Using Frequency Domain ApproachsuryasanNo ratings yet
- 35.full JBC - Enzyme SuperfamiliesDocument8 pages35.full JBC - Enzyme SuperfamiliesmcgilicuttyNo ratings yet
- ArticleDocument11 pagesArticleSahil8No ratings yet
- Article: A de Novo Protein Binding Pair by Computational Design and Directed EvolutionDocument11 pagesArticle: A de Novo Protein Binding Pair by Computational Design and Directed EvolutionSagar KhareNo ratings yet
- Protein complex compositions predicted by structural similarityDocument10 pagesProtein complex compositions predicted by structural similarityhuouinkyoumaNo ratings yet
- Enhancing Ligand Pose Sampling for DockingDocument13 pagesEnhancing Ligand Pose Sampling for DockingalturnerianNo ratings yet
- Target-Oriented Generic Fingerprint-Based Molecular RepresentationDocument14 pagesTarget-Oriented Generic Fingerprint-Based Molecular RepresentationCS & ITNo ratings yet
- A Benchmark Driven Guide To Binding SiteDocument50 pagesA Benchmark Driven Guide To Binding SiteAlexNo ratings yet
- Sequence and Structural Features of Binding Site RDocument7 pagesSequence and Structural Features of Binding Site RAfaque AlamNo ratings yet
- COMPASS: A Tool For Comparison of Multiple Protein Alignments With Assessment of Statistical SignificanceDocument20 pagesCOMPASS: A Tool For Comparison of Multiple Protein Alignments With Assessment of Statistical SignificanceВалерий БжезинскийNo ratings yet
- A Topology-Based Network Tree For The Prediction of Protein-Protein Binding Affinity Changes Following MutationDocument8 pagesA Topology-Based Network Tree For The Prediction of Protein-Protein Binding Affinity Changes Following Mutation1813975862yvyvNo ratings yet
- Bsa CastDocument14 pagesBsa Castaniket singhNo ratings yet
- Babbitt PDFDocument2 pagesBabbitt PDFSamra KanwalNo ratings yet
- 2022 NPJ Syst Biol Appl 8 14Document8 pages2022 NPJ Syst Biol Appl 8 14mbrylinskiNo ratings yet
- 2022 BMC Cancer 22 1211Document17 pages2022 BMC Cancer 22 1211mbrylinskiNo ratings yet
- 2022 Oncotarget 13 695-706Document12 pages2022 Oncotarget 13 695-706mbrylinskiNo ratings yet
- 2019 J Comput Aided Mol Des 33 509-519Document11 pages2019 J Comput Aided Mol Des 33 509-519mbrylinskiNo ratings yet
- 2020 Bioinformatics 36 3077-3083Document7 pages2020 Bioinformatics 36 3077-3083mbrylinskiNo ratings yet
- 2019 BMC Pharmacol Toxicol 20 2Document15 pages2019 BMC Pharmacol Toxicol 20 2mbrylinskiNo ratings yet
- 2019 Biomolecules 9 603Document20 pages2019 Biomolecules 9 603mbrylinskiNo ratings yet
- Pocket2Drug: An Encoder-Decoder Deep Neural Network For The Target-Based Drug DesignDocument12 pagesPocket2Drug: An Encoder-Decoder Deep Neural Network For The Target-Based Drug DesignmbrylinskiNo ratings yet
- 2019 J Mol Graph Model 87 227-239Document13 pages2019 J Mol Graph Model 87 227-239mbrylinskiNo ratings yet
- 2019 Brief Bioinform 20 2167-2184Document18 pages2019 Brief Bioinform 20 2167-2184mbrylinskiNo ratings yet
- 2019 PLoS Comput Biol 15 E1006718Document23 pages2019 PLoS Comput Biol 15 E1006718mbrylinskiNo ratings yet
- 2016 Curr Drug Targets 17 1595-1609Document15 pages2016 Curr Drug Targets 17 1595-1609mbrylinskiNo ratings yet
- 2018 GigaScience 7 1-9Document9 pages2018 GigaScience 7 1-9mbrylinskiNo ratings yet
- 2019 Sci Rep 9 14625Document11 pages2019 Sci Rep 9 14625mbrylinskiNo ratings yet
- 2017 J Virol 91 E00873-17Document14 pages2017 J Virol 91 E00873-17mbrylinskiNo ratings yet
- 2018 Chem Biol Drug Des 91 380-390Document11 pages2018 Chem Biol Drug Des 91 380-390mbrylinskiNo ratings yet
- 2018 J Mol Biol 430 2266-2273Document8 pages2018 J Mol Biol 430 2266-2273mbrylinskiNo ratings yet
- 2017 J Chem Inf Model 57 627-31Document5 pages2017 J Chem Inf Model 57 627-31mbrylinskiNo ratings yet
- 2017 BMC Bioinformatics 18 257Document14 pages2017 BMC Bioinformatics 18 257mbrylinskiNo ratings yet
- 2018 BMC Bioinformatics 19 91Document17 pages2018 BMC Bioinformatics 19 91mbrylinskiNo ratings yet
- 2016 Biochemistry 55 3204-13Document10 pages2016 Biochemistry 55 3204-13mbrylinskiNo ratings yet
- 2016 Comput Biol Chem 64 403-13Document11 pages2016 Comput Biol Chem 64 403-13mbrylinskiNo ratings yet
- 2017 Methods Mol Biol 1611 109-22Document14 pages2017 Methods Mol Biol 1611 109-22mbrylinskiNo ratings yet
- 2018 NPJ Syst Biol Appl 4 13Document10 pages2018 NPJ Syst Biol Appl 4 13mbrylinskiNo ratings yet
- 2016 J Virol 90 10351-61Document11 pages2016 J Virol 90 10351-61mbrylinskiNo ratings yet
- 2016 PLOS ONE 11 E0158898Document29 pages2016 PLOS ONE 11 E0158898mbrylinskiNo ratings yet
- 2016 Bioinformatics 32 579-586Document8 pages2016 Bioinformatics 32 579-586mbrylinskiNo ratings yet
- Methods: Surabhi Maheshwari, Michal BrylinskiDocument8 pagesMethods: Surabhi Maheshwari, Michal BrylinskimbrylinskiNo ratings yet
- 2016 J Virol 90 2230-9Document10 pages2016 J Virol 90 2230-9mbrylinskiNo ratings yet
- 2016 J Cheminform 8 14Document16 pages2016 J Cheminform 8 14mbrylinskiNo ratings yet
- Reverse PharmacognosyDocument15 pagesReverse PharmacognosyJesús GutiérrezNo ratings yet
- NutrientsDocument15 pagesNutrientsFajri FebrianNo ratings yet
- Practise School - ChemistryDocument64 pagesPractise School - ChemistrySonakshi BhatiaNo ratings yet
- UsingAutoDock4forVirtualScreening v4Document37 pagesUsingAutoDock4forVirtualScreening v4Prateek TandonNo ratings yet
- Rational Drug Design Methods Gadavala SarahDocument25 pagesRational Drug Design Methods Gadavala SarahSejal khuman100% (1)
- Muegge 2010Document46 pagesMuegge 2010Santi SuronoNo ratings yet
- Synthetic Drugs in TBDocument46 pagesSynthetic Drugs in TBTausif AlamNo ratings yet
- Instadock: A Single-Click Graphical User Interface For Molecular Docking-Based Virtual High-Throughput ScreeningDocument8 pagesInstadock: A Single-Click Graphical User Interface For Molecular Docking-Based Virtual High-Throughput ScreeningJoan Petit GrosNo ratings yet
- Journal of Ayurveda and Integrative MedicineDocument8 pagesJournal of Ayurveda and Integrative MedicineDrzakir HussainNo ratings yet
- Acs Jcim 2c01400Document8 pagesAcs Jcim 2c01400谢吴辰No ratings yet
- Design of Drug Like Hepsin Inhibitors Against Prostat - 2020 - Acta PharmaceuticDocument12 pagesDesign of Drug Like Hepsin Inhibitors Against Prostat - 2020 - Acta PharmaceuticMohammed Shuaib AhmedNo ratings yet
- Sequence-Based Drug Design As A Concept in Computational Drug DesignDocument21 pagesSequence-Based Drug Design As A Concept in Computational Drug DesignAntonio Aguilar PuenteNo ratings yet
- Pharmacophore Based Drug Design Approach As A Practical Process in Drug DiscoveryDocument13 pagesPharmacophore Based Drug Design Approach As A Practical Process in Drug DiscoverySANJAY K GOWDANo ratings yet
- MolecularDockingStructureBasedDrugDesignStrategies PDFDocument38 pagesMolecularDockingStructureBasedDrugDesignStrategies PDFIvt Morales100% (1)
- Chapter Proposal Submission FormDocument3 pagesChapter Proposal Submission FormLavanya Priya SathyanNo ratings yet
- Docking and ScoringDocument54 pagesDocking and Scoringمحمد حسین لطفیNo ratings yet
- In Silico Study of Agaricus Bisporus On DNA Damaging ProteinDocument7 pagesIn Silico Study of Agaricus Bisporus On DNA Damaging ProteinEditor IJTSRDNo ratings yet
- Custom Scoring CDPK1 With SminaDocument14 pagesCustom Scoring CDPK1 With Sminawadelkrid1270No ratings yet
- AutoDock Vina ManualDocument6 pagesAutoDock Vina ManualBruno AndradeNo ratings yet
- Autodock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python BindingsDocument8 pagesAutodock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python BindingsJoan Petit GrosNo ratings yet
- Biocatalysis and Agricultural Biotechnology: Rakesh Kumar Paul, Virendra Nath, Vipin KumarDocument7 pagesBiocatalysis and Agricultural Biotechnology: Rakesh Kumar Paul, Virendra Nath, Vipin KumarMario Andres JuradoNo ratings yet
- Vuorinen2015 Methods N Apply PharmacophoreDocument22 pagesVuorinen2015 Methods N Apply PharmacophoreRIKANo ratings yet
- Molecular Docking Study of Aspirin and Aspirin DerivativesDocument7 pagesMolecular Docking Study of Aspirin and Aspirin DerivativesInternational Organization of Scientific Research (IOSR)No ratings yet
- AUDocker LE Manual - Virtual Screening Software GuideDocument11 pagesAUDocker LE Manual - Virtual Screening Software GuidedehammoNo ratings yet
- Drug Design - PharmacophoreDocument24 pagesDrug Design - PharmacophoreFatma HishamNo ratings yet
- Role of Computer-Aided Drug Design in Modern Drug DiscoveryDocument16 pagesRole of Computer-Aided Drug Design in Modern Drug DiscoverySourav MajumdarNo ratings yet
- Molecular Dynamics Simulations and Novel Drug DiscoveryDocument16 pagesMolecular Dynamics Simulations and Novel Drug DiscoveryRishavNo ratings yet
- De Souza Neto Et Al. - 2020 - in Silico Strategies To Support Fragment-to-Lead Optimization in Drug DiscoveryDocument18 pagesDe Souza Neto Et Al. - 2020 - in Silico Strategies To Support Fragment-to-Lead Optimization in Drug DiscoveryMritunjay Singh YadavNo ratings yet
- Molecular Docking in Structure-based Drug DesignDocument33 pagesMolecular Docking in Structure-based Drug DesignIvan Tubert-BrohmanNo ratings yet
- Practice School On Research and Development: Key HighlightsDocument17 pagesPractice School On Research and Development: Key Highlightssubhankar pradhanNo ratings yet