You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5806)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (589)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Part 1-Dietary Analysis AssignmentDocument2 pagesPart 1-Dietary Analysis AssignmentHira NasirNo ratings yet
- Marketing AssignmentDocument13 pagesMarketing AssignmentHawe MesfinNo ratings yet
- Vendor Air Compressor Instruction Manual: Nex-A Series Nex A SeriesDocument59 pagesVendor Air Compressor Instruction Manual: Nex-A Series Nex A Seriescraponzel100% (2)
- KAHLIDr Yehia45 PDFDocument36 pagesKAHLIDr Yehia45 PDFLuiz Rubens Souza CantelliNo ratings yet
- Laraya v. Pe, CA-GR No. SP 80927, February 4, 2005Document7 pagesLaraya v. Pe, CA-GR No. SP 80927, February 4, 2005Jonna Maye Loras CanindoNo ratings yet
- Videos On Drug Abuse. Guidelines For Finals Quiz 3 - 5Document1 pageVideos On Drug Abuse. Guidelines For Finals Quiz 3 - 5Gerald John PazNo ratings yet
- Problem Based Learning PDFDocument1 pageProblem Based Learning PDFGerald John PazNo ratings yet
- Phlebotomy: Phlebos - Greek Word Meaning "Vein" & Tome - "To Cut"Document5 pagesPhlebotomy: Phlebos - Greek Word Meaning "Vein" & Tome - "To Cut"Gerald John PazNo ratings yet
- 1 s2.0 S0167701216302627 MainDocument8 pages1 s2.0 S0167701216302627 MainGerald John PazNo ratings yet
- Gene Therapy May Prevent Retinoschisis in Boys: ReviewDocument2 pagesGene Therapy May Prevent Retinoschisis in Boys: ReviewGerald John PazNo ratings yet
- Case Studies On CoagulationDocument3 pagesCase Studies On CoagulationGerald John PazNo ratings yet
- Different Laboratory Service ModelsDocument1 pageDifferent Laboratory Service ModelsGerald John PazNo ratings yet
- Role of Microbiota in Immunity and InflammationDocument12 pagesRole of Microbiota in Immunity and InflammationGerald John PazNo ratings yet
- Biobanking For Human Microbiome Research Promise, Risks, and EthicsDocument15 pagesBiobanking For Human Microbiome Research Promise, Risks, and EthicsGerald John PazNo ratings yet
- Geopolitical Challenges For Infectious Disease Prevention and ControlDocument17 pagesGeopolitical Challenges For Infectious Disease Prevention and ControlGerald John PazNo ratings yet
- Host Microbiomes and Their Interactions With Immune SystemDocument9 pagesHost Microbiomes and Their Interactions With Immune SystemGerald John PazNo ratings yet
- Chapter 023Document34 pagesChapter 023Gerald John PazNo ratings yet
- Chapter 023Document34 pagesChapter 023Gerald John PazNo ratings yet
- GROUP 6-Micro Biochem ResultsDocument2 pagesGROUP 6-Micro Biochem ResultsGerald John PazNo ratings yet
- Chapter 002Document68 pagesChapter 002Gerald John PazNo ratings yet
- Chapter 024Document49 pagesChapter 024Gerald John PazNo ratings yet
- Group 6: Ilagan, Maikka Lopez, Kamla Paz, Gerald John A. Sales, MichelleDocument5 pagesGroup 6: Ilagan, Maikka Lopez, Kamla Paz, Gerald John A. Sales, MichelleGerald John PazNo ratings yet
- RBC Pathology - Part 3Document27 pagesRBC Pathology - Part 3Gerald John PazNo ratings yet
- GROUP 6 MT1031-Micro Biochem ResultsDocument1 pageGROUP 6 MT1031-Micro Biochem ResultsGerald John PazNo ratings yet
- Handwashing Mouth Pipetting Centrifuge Fume Hood Lab Equipment Ppe Reagent Dropper Bottle Reagent Stopper Bottle Ringstand With Wire GauzeDocument2 pagesHandwashing Mouth Pipetting Centrifuge Fume Hood Lab Equipment Ppe Reagent Dropper Bottle Reagent Stopper Bottle Ringstand With Wire GauzeGerald John PazNo ratings yet
- Graves DiseaseDocument13 pagesGraves DiseaseGerald John PazNo ratings yet
- Conn'S Syndrome: Behrens, Aleah Nestine Lagman, Bunny Dawn Perez, Patria NiñaDocument9 pagesConn'S Syndrome: Behrens, Aleah Nestine Lagman, Bunny Dawn Perez, Patria NiñaGerald John PazNo ratings yet
- Laxman Logistics - WriteupDocument7 pagesLaxman Logistics - WriteupGagandeep PahwaNo ratings yet
- Material Safety Data Sheet: SALP Double Acting Baking Powder (Item 112812) Section I - IdentificationDocument2 pagesMaterial Safety Data Sheet: SALP Double Acting Baking Powder (Item 112812) Section I - IdentificationDidier GómezNo ratings yet
- JK Accounts ResearchDocument7 pagesJK Accounts ResearchkrunalNo ratings yet
- Topic: Restaurants: Listening Gap FillDocument2 pagesTopic: Restaurants: Listening Gap FillKim DungNo ratings yet
- List of DOS CommandsDocument22 pagesList of DOS CommandsVikash SharmaNo ratings yet
- Lrfdcons 3 I6Document7 pagesLrfdcons 3 I6diablopapanatasNo ratings yet
- Honesty in Financial DealingDocument4 pagesHonesty in Financial DealingAbdul BasithNo ratings yet
- SGCL Technology OverviewDocument25 pagesSGCL Technology OverviewRajahVKNo ratings yet
- Impedance Stabilization Network (Isn) For Unscreened Balanced PairsDocument3 pagesImpedance Stabilization Network (Isn) For Unscreened Balanced PairsenticoNo ratings yet
- Dede JuleoDocument2 pagesDede JuleoStanindo ArthalanggengNo ratings yet
- Local Council Crisis PlanDocument18 pagesLocal Council Crisis PlanJgjbfhbbgbNo ratings yet
- Which of The Following Falls Under The Activity-Based Management Umbrella?Document17 pagesWhich of The Following Falls Under The Activity-Based Management Umbrella?naddieNo ratings yet
- Chapter 27 MachineryDocument20 pagesChapter 27 MachineryKendall JennerNo ratings yet
- WACC Berger Paints Bahar RoyDocument12 pagesWACC Berger Paints Bahar RoyBahar RoyNo ratings yet
- Lattice eARC WP FINALDocument9 pagesLattice eARC WP FINALSovi SoviNo ratings yet
- DOTr DO No. 2015-011 PDFDocument6 pagesDOTr DO No. 2015-011 PDFEarvin James AtienzaNo ratings yet
- 0 - Oracle Full & SQL TopicDocument168 pages0 - Oracle Full & SQL TopicDivili PadmavathiNo ratings yet
- Sample of Income Statement CertificatesDocument3 pagesSample of Income Statement CertificatesUmar Shahzad JanjuaNo ratings yet
- License Plate Recognition Methods Employing Neural NetworksDocument34 pagesLicense Plate Recognition Methods Employing Neural Networksmuhammad rohmattullahNo ratings yet
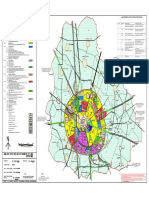
- Jhajjar Master PlanDocument1 pageJhajjar Master PlanMayank AhujaNo ratings yet
- GPPB Circular No. 01-2020Document10 pagesGPPB Circular No. 01-2020Bruce DoyaoenNo ratings yet
- Merging of RC5 With AES - Incorporating More Flexibility and Security To AESDocument6 pagesMerging of RC5 With AES - Incorporating More Flexibility and Security To AESSuhas ManangiNo ratings yet
- 7615 Ijcsit 08Document10 pages7615 Ijcsit 08Anonymous Gl4IRRjzNNo ratings yet
- C-5 Afto 781 GuideDocument15 pagesC-5 Afto 781 GuideRichard Lund100% (1)
- (WWW - Entrance-Exam - Net) - Siemens Placement Sample Paper 2Document6 pages(WWW - Entrance-Exam - Net) - Siemens Placement Sample Paper 2sauravlodhNo ratings yet