You might also like
- Informe de HioscinaDocument9 pagesInforme de HioscinaJosé Ambrosio VillazanaNo ratings yet
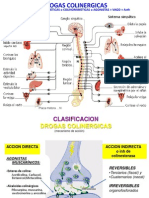
- Drogas ColinergicasDocument49 pagesDrogas Colinergicasnicodemo946No ratings yet
- Alteraciones de Los AminoacidosDocument9 pagesAlteraciones de Los AminoacidosAlejandro Rodas SalinasNo ratings yet
- Actividad 01 - S2 - Farmacocinética - FarmacodinámicaDocument18 pagesActividad 01 - S2 - Farmacocinética - FarmacodinámicaOlgaMuñozChamayaNo ratings yet
- Unión NeuromuscularDocument9 pagesUnión NeuromuscularjaimeNo ratings yet
- Efectores Alostéricos Positivos y Negativos Regulan Enzimas ClaveDocument13 pagesEfectores Alostéricos Positivos y Negativos Regulan Enzimas ClaveGrecia Vera VallejosNo ratings yet
- FurfuralDocument6 pagesFurfuralDaniel CoronadoNo ratings yet
- Antagonistas de Receptores H2 DOCUMENTODocument6 pagesAntagonistas de Receptores H2 DOCUMENTOAlex Renteria AparicioNo ratings yet
- Tarea 6 TOXIDocument2 pagesTarea 6 TOXIMarisol CGNo ratings yet
- Te VerdeDocument6 pagesTe VerdeRosángela RodríguezNo ratings yet
- Marco TeoricoDocument8 pagesMarco TeoricoCristopherEsquivelSaavedraNo ratings yet
- SINTESIS DE COLESTEROL ResumenDocument4 pagesSINTESIS DE COLESTEROL ResumenXena Katherine Gutarra SosaNo ratings yet
- Farmacos Moduladores de La NoradrenalinaDocument5 pagesFarmacos Moduladores de La NoradrenalinaJazmin Shany StellaNo ratings yet
- El ciclo de la urea: transformación del amoníaco en ureaDocument65 pagesEl ciclo de la urea: transformación del amoníaco en ureaAlexandraMedinaCastilloNo ratings yet
- Practica 10 NicotinaDocument21 pagesPractica 10 NicotinaVan_der_WaalsNo ratings yet
- NicotinaDocument11 pagesNicotinaanaNo ratings yet
- Requisitos y clasificación de excipientes farmacéuticosDocument10 pagesRequisitos y clasificación de excipientes farmacéuticosMilagros Santamaría InoñanNo ratings yet
- Cocos Gram Negativos (Neisserias)Document2 pagesCocos Gram Negativos (Neisserias)aaronlawlietNo ratings yet
- Reporte de Colesterol Total y TriglicéridosDocument6 pagesReporte de Colesterol Total y TriglicéridosjeiselllaNo ratings yet
- Enfermedad de McArdleDocument23 pagesEnfermedad de McArdleJaimeD.LópezAlcalá100% (1)
- Metabolismo de glucosa, fructuosa y galactosaDocument2 pagesMetabolismo de glucosa, fructuosa y galactosaKarol MontañoNo ratings yet
- Farmacología Adrenérgica y ColinérgicaDocument6 pagesFarmacología Adrenérgica y ColinérgicaJhon AnccellNo ratings yet
- Practica 6 Analisis de Vitaminas y MineralesDocument5 pagesPractica 6 Analisis de Vitaminas y MineralesLuis BahachilleNo ratings yet
- Glucogeno Tipo V (Mcardie)Document12 pagesGlucogeno Tipo V (Mcardie)Edgar Omar Campos MartinezNo ratings yet
- Heterósidos Antraquinónicos Clase 5Document26 pagesHeterósidos Antraquinónicos Clase 5Belén LuqueNo ratings yet
- PRACTICA 5 CUESTIONARIO Velocidad de DisolucionDocument3 pagesPRACTICA 5 CUESTIONARIO Velocidad de DisolucionAxel .iNo ratings yet
- Seminario 5 - Fisiología Endocrina 1Document9 pagesSeminario 5 - Fisiología Endocrina 1Fernanda Elizabeth MolinaNo ratings yet
- Temas 11 y 12 - Lipidos FarmaciaDocument29 pagesTemas 11 y 12 - Lipidos FarmaciaGrisel RodriguezNo ratings yet
- Personal en La Farmacia PDFDocument26 pagesPersonal en La Farmacia PDFMarco Vargas VNo ratings yet
- Intox - ParacetamolDocument31 pagesIntox - ParacetamolJhon GomezNo ratings yet
- Alplax Net G00073201 07Document6 pagesAlplax Net G00073201 07Matias TomasettoNo ratings yet
- ZIMÓGENOSDocument13 pagesZIMÓGENOSLuiggi GordilloNo ratings yet
- Clase 09 Farmacologia BronquialDocument26 pagesClase 09 Farmacologia Bronquialjuana milagros hidalgoaedoNo ratings yet
- E6. Farmacología Ii AntiviricosDocument19 pagesE6. Farmacología Ii AntiviricosDania Baculima100% (1)
- AMITRIPTILINADocument6 pagesAMITRIPTILINABilly Reyna BlasNo ratings yet
- Cardiotónicos y contractilidad cardíacaDocument24 pagesCardiotónicos y contractilidad cardíacaWillian FernandezNo ratings yet
- 16cfarmacologaadrenrgicos 110829023054 Phpapp01Document114 pages16cfarmacologaadrenrgicos 110829023054 Phpapp01Guardia ItebaNo ratings yet
- Alcaloides Derivados de La Fenilalanina y TirosinaDocument14 pagesAlcaloides Derivados de La Fenilalanina y TirosinaSergio Llontop OrdoñezNo ratings yet
- Receptores de GlicinaDocument3 pagesReceptores de GlicinaOscar CáceresNo ratings yet
- Heterosidos CianogeneticosDocument26 pagesHeterosidos CianogeneticosVileanNo ratings yet
- Alcaloides Del Ergot 10..3..2014Document3 pagesAlcaloides Del Ergot 10..3..2014Jos Leon Castillo Arroyave100% (1)
- Apaza Solano Harold DanielPractica N°1 ResolucionDocument8 pagesApaza Solano Harold DanielPractica N°1 ResolucionDaniel Apaza SolanoNo ratings yet
- Metabolismoi de AminiacidosDocument7 pagesMetabolismoi de Aminiacidosanon_609905353No ratings yet
- HaptenoDocument6 pagesHaptenoAnira Angel Aguilar AguilarNo ratings yet
- Efedrina: Monografía sobre sus propiedades químicas y usos terapéuticosDocument2 pagesEfedrina: Monografía sobre sus propiedades químicas y usos terapéuticosMauricio RodríguezNo ratings yet
- GUIAS SEMINARIOS FARMACO 2 SEMINARIO No 7Document3 pagesGUIAS SEMINARIOS FARMACO 2 SEMINARIO No 7Maria100% (1)
- Farmacologia GanglionarDocument7 pagesFarmacologia GanglionarjeniferNo ratings yet
- Biosintesis TyrDocument1 pageBiosintesis TyrAndreziithoMgNo ratings yet
- Seminario 2Document4 pagesSeminario 2Grisell TorresNo ratings yet
- Practica 2....Document4 pagesPractica 2....Nei Marllori Davila CaisaraNo ratings yet
- Sintesis TAMOXIFENODocument12 pagesSintesis TAMOXIFENODayana MoralesNo ratings yet
- Deficiencia de asparagina sintetasa, pruebas genéticas ASNSDocument2 pagesDeficiencia de asparagina sintetasa, pruebas genéticas ASNSDariforiNo ratings yet
- Determinacion Cuantitativa Del Acido UricoDocument14 pagesDeterminacion Cuantitativa Del Acido UricoPedro SuarezNo ratings yet
- Tranquilizantes MayoresDocument65 pagesTranquilizantes MayoresMonse RodriguezNo ratings yet
- Labo 10 GnosiaDocument4 pagesLabo 10 GnosiaGrecia Jaime HerreraNo ratings yet
- Practica de AnestesicosDocument22 pagesPractica de AnestesicosDiana Sofía Villanueva YaveNo ratings yet
- Farmacologia A LuenaDocument164 pagesFarmacologia A LuenaPedro Emilio LeonNo ratings yet
- Farmacocinética y administración de dosis múltiplesDocument2 pagesFarmacocinética y administración de dosis múltiplesemmgrNo ratings yet
- Biotensis de Los Aminoacidos No Esenciales Tema 10Document9 pagesBiotensis de Los Aminoacidos No Esenciales Tema 10Richard PalaciosNo ratings yet
- Deficiencia de Carnitina: Caso de un Paciente con Síndrome HipotónicoDocument11 pagesDeficiencia de Carnitina: Caso de un Paciente con Síndrome HipotónicoRodriguezMarielleNo ratings yet
- Bombas Efervescentes para PiesDocument5 pagesBombas Efervescentes para PiesAladino AbbasNo ratings yet
- Keratina para El CabelloDocument5 pagesKeratina para El CabelloAladino AbbasNo ratings yet
- Estética de UñasDocument4 pagesEstética de UñasAladino AbbasNo ratings yet
- AlquimiaDocument22 pagesAlquimiaAladino AbbasNo ratings yet
- Estructura de La Uña Natural y Anatomía de La UñaDocument8 pagesEstructura de La Uña Natural y Anatomía de La UñaAladino AbbasNo ratings yet
- Orbital AtómicoDocument5 pagesOrbital AtómicoAladino AbbasNo ratings yet
- ParoniquiaDocument3 pagesParoniquiaAladino AbbasNo ratings yet
- El Mejor Monómero para Uñas AcrílicasDocument5 pagesEl Mejor Monómero para Uñas AcrílicasAladino AbbasNo ratings yet
- Uñas AcrílicasDocument8 pagesUñas AcrílicasAladino AbbasNo ratings yet
- Padrastro (Uña)Document1 pagePadrastro (Uña)Aladino AbbasNo ratings yet
- AlquimiaDocument22 pagesAlquimiaAladino AbbasNo ratings yet
- La AlquimiaDocument7 pagesLa AlquimiaAladino AbbasNo ratings yet
- Neutrón 4Document4 pagesNeutrón 4Aladino AbbasNo ratings yet
- AlquimiaDocument22 pagesAlquimiaAladino AbbasNo ratings yet
- Átomo 1Document14 pagesÁtomo 1Aladino AbbasNo ratings yet
- Electrón 2Document30 pagesElectrón 2Aladino AbbasNo ratings yet
- HematuriaDocument8 pagesHematuriaAladino Abbas100% (1)
- Protón 3Document5 pagesProtón 3Aladino AbbasNo ratings yet
- Análisis de Sedimentos en La OrinaDocument5 pagesAnálisis de Sedimentos en La OrinaAladino AbbasNo ratings yet
- Biofísica WikiDocument4 pagesBiofísica WikiAladino AbbasNo ratings yet
- Gregor MendelDocument8 pagesGregor MendelAladino AbbasNo ratings yet
- Biología WikiDocument15 pagesBiología WikiAladino AbbasNo ratings yet
- Ga LactosaDocument2 pagesGa LactosaAladino AbbasNo ratings yet
- 09 El BigbangDocument196 pages09 El BigbangRey LagartoNo ratings yet
- Lactasa PDFDocument6 pagesLactasa PDFAladino AbbasNo ratings yet
- Lactasa PDFDocument6 pagesLactasa PDFAladino AbbasNo ratings yet
- 09 El BigbangDocument196 pages09 El BigbangRey LagartoNo ratings yet
- Galactose MiaDocument10 pagesGalactose MiaAladino AbbasNo ratings yet
- Lactasa PDFDocument6 pagesLactasa PDFAladino AbbasNo ratings yet
- LactosaDocument4 pagesLactosaAladino AbbasNo ratings yet
- Codigo GeneticoDocument4 pagesCodigo GeneticoyomarNo ratings yet
- ChatDocument164 pagesChatCarlo BrandtNo ratings yet
- Transcripcion y TraduccionDocument64 pagesTranscripcion y TraduccionFABIANA ANDREA DAZA ROCHANo ratings yet
- Tesis PDFDocument88 pagesTesis PDFMerced Baneza CahuanaNo ratings yet
- Compilado BC 2021Document47 pagesCompilado BC 2021Sofi VidalNo ratings yet
- Material didáctico Biología MolecularDocument46 pagesMaterial didáctico Biología MolecularJosé Luis FranzoniNo ratings yet
- Actividad Pág#26 y Resumen (26-30) Biología Aracely RodríguezDocument5 pagesActividad Pág#26 y Resumen (26-30) Biología Aracely RodríguezBryanNo ratings yet
- Genética: transcripción y traducciónDocument25 pagesGenética: transcripción y traducciónYhadira ChinoNo ratings yet
- Icfes Bachillerato 9 GradoDocument31 pagesIcfes Bachillerato 9 GradoLuis EstradaNo ratings yet
- Resumo Biologia MolecularDocument31 pagesResumo Biologia MolecularJOAO ARISTIDES RAMOSNo ratings yet
- Batería Preguntas Tests RafaDocument14 pagesBatería Preguntas Tests Rafatopipa vato tovaNo ratings yet
- Replicacion Taller de BiologiaDocument17 pagesReplicacion Taller de BiologiaDANIA CAROLINA GUTIERREZ HINOJOSA estNo ratings yet
- Resumen de VideosDocument3 pagesResumen de Videosann Go GaNo ratings yet
- Codones de TerminoDocument79 pagesCodones de TerminoYazmin OsorioNo ratings yet
- Socialización Unidad Biología MolecularDocument26 pagesSocialización Unidad Biología MolecularAlejandraNo ratings yet
- rEPLICACION, TRANSP TRADDocument8 pagesrEPLICACION, TRANSP TRADConsuelo UlloaNo ratings yet
- Guía Grado 9°Document17 pagesGuía Grado 9°SANDRA MILENA AYALA LENISNo ratings yet
- Taller 3 HECHO PDFDocument7 pagesTaller 3 HECHO PDFPascual FerrerNo ratings yet
- ADN y ARN: estructura y funcionesDocument22 pagesADN y ARN: estructura y funcionesLuna Hernández Adamaris MonserratNo ratings yet
- OperonesDocument8 pagesOperonesMarisol Montero LopezNo ratings yet
- Taller Sintesis de ProteinasDocument1 pageTaller Sintesis de Proteinasanon_37704526No ratings yet
- Proceso de Maduracion de Los Diferentes ArnDocument4 pagesProceso de Maduracion de Los Diferentes ArnJosselynSánchezYRichardBarrezuetaNo ratings yet
- 02 El Genoma HumanoDocument25 pages02 El Genoma HumanogilbertomartinezNo ratings yet
- Elvia Isabel Guacaneme Perez, Taller 5Document4 pagesElvia Isabel Guacaneme Perez, Taller 5Elvia Isabel GUACANEME PEREZNo ratings yet
- Genetica Bacteriana e Introduccion Al Dna RecombinanteDocument11 pagesGenetica Bacteriana e Introduccion Al Dna RecombinanteJinel MendozaNo ratings yet
- La apigenina, la kriptonita del cáncerDocument3 pagesLa apigenina, la kriptonita del cáncertito cuadrosNo ratings yet
- Expresion Genica, Transcripcion y TraduccionDocument27 pagesExpresion Genica, Transcripcion y TraduccionJesus ruiz bacaNo ratings yet
- ARNtDocument52 pagesARNtLiza Andrea Allende LópezNo ratings yet
- FISIOLOGÍA 1 TRABAJO 1 AndreaDocument13 pagesFISIOLOGÍA 1 TRABAJO 1 AndreaSUSAN ANDREA DORADONo ratings yet
- Examen II ParcialDocument16 pagesExamen II ParcialMauricio GalvezNo ratings yet