You might also like
- Structure Determination Problems Using Ir SpectrosDocument5 pagesStructure Determination Problems Using Ir SpectrosSaurav PaulNo ratings yet
- Molecular Docking and 3D-QSAR Based Design ofDocument12 pagesMolecular Docking and 3D-QSAR Based Design ofJ-Paul DétoNo ratings yet
- Roughness Conversion Chart RBTDocument1 pageRoughness Conversion Chart RBTMalwadkar P.B.No ratings yet
- The Environment (Protection) Amendment Rules, 2008Document9 pagesThe Environment (Protection) Amendment Rules, 2008Krutika BhatNo ratings yet
- Cyclohexane 1,3 DioneDocument1 pageCyclohexane 1,3 DionetreemaddogNo ratings yet
- Entry Ar Mol - WT (G/mole) 330.35: PH O N H N O BRDocument2 pagesEntry Ar Mol - WT (G/mole) 330.35: PH O N H N O BRHamza AssilaNo ratings yet
- Selected Total Syntheses (Furstner) PDFDocument280 pagesSelected Total Syntheses (Furstner) PDFludoNo ratings yet
- Carboxylic AcidDocument16 pagesCarboxylic AcidArnav SainiNo ratings yet
- Figure 1Document1 pageFigure 1MRITZNo ratings yet
- Img 20200511 0001Document1 pageImg 20200511 0001Bashar Al AliNo ratings yet
- USBASP 51/AVR编程器: 5V Vcc2Board Mosi RST SCK MisoDocument1 pageUSBASP 51/AVR编程器: 5V Vcc2Board Mosi RST SCK Misoframqz100% (1)
- 26 TZ 1251994Document43 pages26 TZ 1251994AdeAryadewanataNo ratings yet
- F-350 Steering Gear Hyd DWGDocument1 pageF-350 Steering Gear Hyd DWGisabella bibkoNo ratings yet
- Research ArticleDocument16 pagesResearch ArticleCarla Luna MinucheNo ratings yet
- Arynes in Natural Product SynthesisDocument14 pagesArynes in Natural Product SynthesisIJRASETPublicationsNo ratings yet
- Arynes in Natural Product SynthesisDocument14 pagesArynes in Natural Product SynthesisIJRASETPublicationsNo ratings yet
- Chapter 08 Test ReviewDocument5 pagesChapter 08 Test ReviewКанат ТютеновNo ratings yet
- Motor Procesamiento Optoacoplador Driver V1 5V R1 1kΩ encoder LoadDocument1 pageMotor Procesamiento Optoacoplador Driver V1 5V R1 1kΩ encoder LoadYefferFernandezNo ratings yet
- Jur IyaDocument5 pagesJur IyaNurmuslimahhhNo ratings yet
- SN74LS247Document1 pageSN74LS247Francis GuilbaultNo ratings yet
- BF 00714771Document3 pagesBF 00714771idanfriNo ratings yet
- Exp Teco 2Document4 pagesExp Teco 2sandi suharyadiNo ratings yet
- Omega Lighting L7003-L9004 TW & TWD INC A-Lamp Lensed Downlight Spec Sheet 9-85Document2 pagesOmega Lighting L7003-L9004 TW & TWD INC A-Lamp Lensed Downlight Spec Sheet 9-85Alan MastersNo ratings yet
- SP 9s 5 005 Dgeva Tds TypeDocument2 pagesSP 9s 5 005 Dgeva Tds TypeLINo ratings yet
- AT89S52Document3 pagesAT89S52Gopi NadhNo ratings yet
- KTS Chương 4Document5 pagesKTS Chương 4Trịnh Quốc ThànhNo ratings yet
- Altered Connections On The Road To PsychopathyDocument4 pagesAltered Connections On The Road To PsychopathyAfa ValverdeNo ratings yet
- Tablas de Acidez y de Grupos SalientesDocument6 pagesTablas de Acidez y de Grupos Salientesjuan pablo isaza riosNo ratings yet
- General Chemistry ToolsDocument3 pagesGeneral Chemistry ToolsJA BerzabalNo ratings yet
- 1.6/ S BD-BD (1:2) : No19v 9í't2 BE (1:1)Document1 page1.6/ S BD-BD (1:2) : No19v 9í't2 BE (1:1)Eduardo RodriguezNo ratings yet
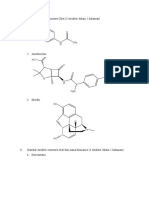
- I. Gambar Struktur Senyawa Obat (3 Struktur Dalam 1 Halaman) 1. ParacetamolDocument4 pagesI. Gambar Struktur Senyawa Obat (3 Struktur Dalam 1 Halaman) 1. ParacetamolFebe HarumNo ratings yet
- Stats QuestionsDocument7 pagesStats QuestionsBansil PatelNo ratings yet
- Up+Trolled Copy: Issued Ay Cts PP PDocument1 pageUp+Trolled Copy: Issued Ay Cts PP PDave ChaudhuryNo ratings yet
- C C Bond Formation: From Other AcetylenesDocument4 pagesC C Bond Formation: From Other AcetylenesrashidNo ratings yet
- EG&D - SHEET 06 - SolidDocument7 pagesEG&D - SHEET 06 - Solidptldarshan22No ratings yet
- Metal Scavanger GuideDocument12 pagesMetal Scavanger Guidebharadwajnarayanam100% (1)
- Sample UkuranDocument2 pagesSample UkuranCici LiantiNo ratings yet
- Sachin Rana (Iitb) : E-Z Double BondsDocument3 pagesSachin Rana (Iitb) : E-Z Double BondsGauransh Goyal 553No ratings yet
- Elion - Quimica ComputacionalDocument8 pagesElion - Quimica ComputacionalAlexhitoo TlvNo ratings yet
- Question 2 PDFDocument1 pageQuestion 2 PDFSaketh BoggavarapuNo ratings yet
- Accepted Manuscript: Applied Thermal EngineeringDocument38 pagesAccepted Manuscript: Applied Thermal Engineeringlollapalooza109_5725No ratings yet
- OK Flux 10.62 (Low-Alloyed Steels)Document4 pagesOK Flux 10.62 (Low-Alloyed Steels)גרבר פליקסNo ratings yet
- Steam Separator Vickers 2Document4 pagesSteam Separator Vickers 2Rizki HermawanNo ratings yet
- Tarea 1 EstaticaDocument3 pagesTarea 1 EstaticaAngel GlNo ratings yet
- Cannabinoid Receptor Ligands ReviewDocument16 pagesCannabinoid Receptor Ligands ReviewAllen MokNo ratings yet
- Caaba: Math 4Document2 pagesCaaba: Math 4api-444439435No ratings yet
- 2 - H-NMRDocument38 pages2 - H-NMRahmed mohamedNo ratings yet
- керам 4Document1 pageкерам 4Georgii DavlianidzeNo ratings yet
- GAS So, O, N2Document1 pageGAS So, O, N2Fiel A'nNo ratings yet
- Chen 2012Document3 pagesChen 2012Jaydeep MokariyaNo ratings yet
- Allanson IgnitionCatalogue English 21-2971321Document28 pagesAllanson IgnitionCatalogue English 21-2971321Giovanni MontesNo ratings yet
- AUDIOMETRYDocument1 pageAUDIOMETRYHumas PemasaranNo ratings yet
- END68634Document1 pageEND68634suraj pandeyNo ratings yet
- Adc0808 Pic16f84aDocument1 pageAdc0808 Pic16f84aPhap NguyenNo ratings yet
- Rare Earth Elements and Spider DiagramDocument10 pagesRare Earth Elements and Spider DiagramWisnu astamanNo ratings yet
- Divergent C H Functionalizations Directed by Sulfonamide Pharmacophores: Late-Stage Diversification As A Tool For Drug DiscoveryDocument7 pagesDivergent C H Functionalizations Directed by Sulfonamide Pharmacophores: Late-Stage Diversification As A Tool For Drug DiscoveryEdwinNo ratings yet
- Reactions of 3 - (4-Aryl-2-Thiazolyl) - and 3 - (2-Benzothiazolyl) - 2-Iminocoumarins With N-NucleophilesDocument8 pagesReactions of 3 - (4-Aryl-2-Thiazolyl) - and 3 - (2-Benzothiazolyl) - 2-Iminocoumarins With N-Nucleophileslabsoa111No ratings yet
- Adobe Scan 27-Feb-2023Document8 pagesAdobe Scan 27-Feb-2023Praveenkumar VNo ratings yet
- Chapter 10Document18 pagesChapter 10Khaled NaseerNo ratings yet
- 12 Chemistry Impq CH07 The P Block Elements 02Document21 pages12 Chemistry Impq CH07 The P Block Elements 02Saurabh PatilNo ratings yet
- FulltextDocument121 pagesFulltextSamuel GarciaNo ratings yet
- Differences Between Organic and Inorganic CompoundsDocument3 pagesDifferences Between Organic and Inorganic CompoundsChristelle LynNo ratings yet
- Chapter 10 Chemical Bonding II Molecular Geometry and PolarityDocument45 pagesChapter 10 Chemical Bonding II Molecular Geometry and PolarityAnge'le Mae CISNERONo ratings yet
- Ch8 NotesDocument14 pagesCh8 NotesTriet NguyenNo ratings yet
- STPM Trials 2009 Chemistry Paper 1 (Pahang)Document12 pagesSTPM Trials 2009 Chemistry Paper 1 (Pahang)Looi Chui YeanNo ratings yet
- RevisDocument28 pagesRevisPrabhakar BandaruNo ratings yet
- CH 8 Ionic CompoundsDocument36 pagesCH 8 Ionic CompoundseherrerahghsNo ratings yet
- Chemistry SS2 EditedDocument150 pagesChemistry SS2 EditedSamuel BiyamaNo ratings yet
- Mark Scheme (Results) : October 2017Document24 pagesMark Scheme (Results) : October 2017Ahmed AmanNo ratings yet
- 2.04 - 2.06 Redox Reactions, Halogens and Alkali Earth Metals MSDocument28 pages2.04 - 2.06 Redox Reactions, Halogens and Alkali Earth Metals MSJames ChongNo ratings yet
- Group 16 ElementsDocument40 pagesGroup 16 Elementstapas kunduNo ratings yet
- GOC TheoryDocument11 pagesGOC Theorygargreyansh830No ratings yet
- GeneralChemistry (Organic Compounds)Document27 pagesGeneralChemistry (Organic Compounds)yen perioles100% (1)
- Sat TestDocument14 pagesSat TestDuy Khang Bui100% (1)
- 1 11 Dent MatDocument33 pages1 11 Dent MatkrstnkyslNo ratings yet
- B.tech CurriculumDocument238 pagesB.tech CurriculumMohammed iliyasNo ratings yet
- Ir SpectrosDocument54 pagesIr SpectrosRutvi shahNo ratings yet
- Chemmatters April2007 PaintballDocument4 pagesChemmatters April2007 PaintballNELIE FRANCE SANCHEZNo ratings yet
- 30 07 2022 JR.C IPL (Incoming) Jee Main WTM 04 Q.paperDocument12 pages30 07 2022 JR.C IPL (Incoming) Jee Main WTM 04 Q.paperMurari MarupuNo ratings yet
- Interactions Between Gelatin and Sodium Alginate: UV and FTIR StudiesDocument11 pagesInteractions Between Gelatin and Sodium Alginate: UV and FTIR StudiesAlyanah AloveraNo ratings yet
- Weekly Home Learning Plan-2nd QuarterDocument4 pagesWeekly Home Learning Plan-2nd Quarterdivina bentayaoNo ratings yet
- CHEMISTRYDocument70 pagesCHEMISTRYF E R N A NNo ratings yet
- AQA CHEM5 W MS Jan11Document15 pagesAQA CHEM5 W MS Jan11fukhrajNo ratings yet
- 1 Grade 11 Review AnswersDocument9 pages1 Grade 11 Review Answersapi-363234558No ratings yet
- Ch. - 1. Structure - and - Bonding Organic. Org 1Document71 pagesCh. - 1. Structure - and - Bonding Organic. Org 1Fawzia AuliaNo ratings yet
- Pharm. Organic Chemistry-Final ExamDocument43 pagesPharm. Organic Chemistry-Final ExamKate MendozaNo ratings yet
- REACTION-MECHANISM HandoutDocument3 pagesREACTION-MECHANISM HandoutAlthea Jones S. SaysayNo ratings yet
- Organic Chemistry 2 Practice Exam 1Document15 pagesOrganic Chemistry 2 Practice Exam 1KaybidoNo ratings yet