You might also like
- Ribosome-inactivating Proteins: Ricin and Related ProteinsFrom EverandRibosome-inactivating Proteins: Ricin and Related ProteinsFiorenzo StirpeNo ratings yet
- Rep Sequences: DiscoveryDocument2 pagesRep Sequences: DiscoverySwati2013No ratings yet
- Anatomy of A GeneDocument33 pagesAnatomy of A GenemskikiNo ratings yet
- Nar00064 0196Document8 pagesNar00064 0196Pipe pelaezNo ratings yet
- Bacterial Expression and Enzymatic Activity Analysis of ME1 A Ribosome-Inactivating Protein From Mirabilis Expansa - USADocument10 pagesBacterial Expression and Enzymatic Activity Analysis of ME1 A Ribosome-Inactivating Protein From Mirabilis Expansa - USAT AmaruNo ratings yet
- pRS415Document12 pagespRS415DiegoNo ratings yet
- Glass y Donaldson, 1994Document8 pagesGlass y Donaldson, 1994sararmentaNo ratings yet
- Gene ExpressionDocument10 pagesGene ExpressionRatu HamidNo ratings yet
- 8 Sigmas2003Document7 pages8 Sigmas2003AsmaNo ratings yet
- Widespread Polycistronic Gene Expression in Green Algae: SignificanceDocument10 pagesWidespread Polycistronic Gene Expression in Green Algae: SignificanceAbhishek SahuNo ratings yet
- Fis Upstream BindingDocument15 pagesFis Upstream BindingDiegoNo ratings yet
- 1323 FullDocument8 pages1323 FullJelle van CampenhoutNo ratings yet
- An Effort To Make Sense of Antisense Transcription in BacteriaDocument6 pagesAn Effort To Make Sense of Antisense Transcription in BacteriaNina HernandezNo ratings yet
- Pnas01519 0334Document6 pagesPnas01519 0334medtubeburnerNo ratings yet
- Effect of Restricted Dissolved Oxygen On Expression of Clostridium Difficile Toxin A Subunit From E. ColiDocument13 pagesEffect of Restricted Dissolved Oxygen On Expression of Clostridium Difficile Toxin A Subunit From E. ColieeeNo ratings yet
- The Bifidobacterial and Lactobacillus Microflora of Humans: Gerald W. TannockDocument23 pagesThe Bifidobacterial and Lactobacillus Microflora of Humans: Gerald W. TannockHime IbaraNo ratings yet
- Session 5Document28 pagesSession 5IndhumathiNo ratings yet
- Bioinformatics: Discovery NoteDocument4 pagesBioinformatics: Discovery NotelaksiyerNo ratings yet
- The rDNA Loci-Intersections of Replication, Transcription, and Repair PathwaysDocument14 pagesThe rDNA Loci-Intersections of Replication, Transcription, and Repair PathwaysBenjamín Milla FructuosoNo ratings yet
- Rapid Identification of Reaction-Amplified Ribosomal Spacer PolymorphismsDocument8 pagesRapid Identification of Reaction-Amplified Ribosomal Spacer PolymorphismsKoirala PrijunNo ratings yet
- Biology PaperDocument7 pagesBiology PaperKaixian LiuNo ratings yet
- Site-Specific Recombination StrategiesDocument9 pagesSite-Specific Recombination StrategiesEdgar AbadiaNo ratings yet
- Article eDocument10 pagesArticle eRachelle BriaNo ratings yet
- Virus 1Document9 pagesVirus 1Novita RindoNo ratings yet
- Conserved RNA Secondary Structures in Genomes: PicornaviridaeDocument11 pagesConserved RNA Secondary Structures in Genomes: Picornaviridaenareman hassanNo ratings yet
- 705 FullDocument10 pages705 FullLulu AzizahNo ratings yet
- Compact HK Genes 2Document8 pagesCompact HK Genes 2Chaman ChutiyaNo ratings yet
- Epigenetic Inheritance in PlantsDocument7 pagesEpigenetic Inheritance in Plantsapi-20009652No ratings yet
- Mojica 2005Document9 pagesMojica 2005Django BoyeeNo ratings yet
- Complete Genomic Structrue of M. Aeruginosa NIES 843. Kaneki 2007Document10 pagesComplete Genomic Structrue of M. Aeruginosa NIES 843. Kaneki 2007lorenaNo ratings yet
- 25483p5CliDY PDFDocument15 pages25483p5CliDY PDFArgadinata MargawatiNo ratings yet
- Molecular Characterization of A 28 Kda Surface Antigen Gene Family of The Tribe EhrlichiaeDocument8 pagesMolecular Characterization of A 28 Kda Surface Antigen Gene Family of The Tribe EhrlichiaeDIANA PATRICIA MOLANO PEREZNo ratings yet
- Split GeneDocument27 pagesSplit Genemanojitchatterjee2007No ratings yet
- BT 301 Short Question Mid TermDocument11 pagesBT 301 Short Question Mid TermAlishba khan100% (1)
- Ponting, Et Al 2009. Evolution and Functions of Long Noncoding RNAs.Document13 pagesPonting, Et Al 2009. Evolution and Functions of Long Noncoding RNAs.J Alberto LucasNo ratings yet
- Enterococcal Genetics: Keith E. WeaverDocument33 pagesEnterococcal Genetics: Keith E. WeaverAnastasija KostićNo ratings yet
- Fines Estrictamente Académicos. Artículo Free de NatureDocument42 pagesFines Estrictamente Académicos. Artículo Free de NatureAmando CasasNo ratings yet
- Identification of CRISPR and Riboswitch Related RNAs Among Novel Noncoding RNAs of The Euryarchaeon Pyrococcus AbyssiDocument15 pagesIdentification of CRISPR and Riboswitch Related RNAs Among Novel Noncoding RNAs of The Euryarchaeon Pyrococcus AbyssianasenNo ratings yet
- Imsd Group 1 FinalDocument1 pageImsd Group 1 Finalapi-249816034No ratings yet
- Content ServerDocument11 pagesContent ServerKeri Gobin SamarooNo ratings yet
- Chapter 5 - Basic Concepts in Human Molecular Geneti - 2009 - Molecular PathologDocument19 pagesChapter 5 - Basic Concepts in Human Molecular Geneti - 2009 - Molecular PathologSELNo ratings yet
- An Evaluation of Terminal Restriction Fragment Length Polymorphsim Analysis For The Study of Microbial Community Structure and DynamicsDocument12 pagesAn Evaluation of Terminal Restriction Fragment Length Polymorphsim Analysis For The Study of Microbial Community Structure and DynamicsWilmer HerreraNo ratings yet
- Dynamics MeaningDocument13 pagesDynamics MeaningJay Prakash MauryaNo ratings yet
- Vida Criada em LaboratórioDocument10 pagesVida Criada em LaboratórioCampos VictorNo ratings yet
- Rupak PartDocument174 pagesRupak PartAnup HalderNo ratings yet
- Mechanisms of Long Noncoding RNA Function in Development and DiseaseDocument19 pagesMechanisms of Long Noncoding RNA Function in Development and DiseaseManuel HernándezNo ratings yet
- Sci PaperDocument11 pagesSci PapercalumNo ratings yet
- Assignment of GeneticsDocument8 pagesAssignment of GeneticsSalma AymanNo ratings yet
- Cladistic Relationships Among The Pleurotus Ostreatus Complex, The Pleurotus Pulmonarius Complex, and Pleurotus Eryngii Based On The Mitochondrial Small Subunit Ribosomal DNA Sequence AnalysisDocument6 pagesCladistic Relationships Among The Pleurotus Ostreatus Complex, The Pleurotus Pulmonarius Complex, and Pleurotus Eryngii Based On The Mitochondrial Small Subunit Ribosomal DNA Sequence AnalysisAlejandro Murillo V.No ratings yet
- Apicomplexan AP2Document13 pagesApicomplexan AP2somasushmaNo ratings yet
- DNA Primers For Amplification of Mitochondrial Cytochrome C Oxidase Subunit I From Diverse Metazoan InvertebratesDocument6 pagesDNA Primers For Amplification of Mitochondrial Cytochrome C Oxidase Subunit I From Diverse Metazoan InvertebratesxuanchaiNo ratings yet
- The Complete Chloroplast DNA Sequence of Eleutherococcus Senticosus (Araliaceae) Comparative Evolutionary Analyses With Other Three AsteridsDocument12 pagesThe Complete Chloroplast DNA Sequence of Eleutherococcus Senticosus (Araliaceae) Comparative Evolutionary Analyses With Other Three AsteridsOanaNo ratings yet
- 18 BorowiecDocument30 pages18 Borowiec2begeniusNo ratings yet
- A Complete Immunoglobulin Gene Is Created by Somatic RecombinatioDocument14 pagesA Complete Immunoglobulin Gene Is Created by Somatic Recombinatiomaxence tricaudNo ratings yet
- Adaptive Evolution and Functional Divergence of Pepsin Gene FamilyDocument10 pagesAdaptive Evolution and Functional Divergence of Pepsin Gene FamilyharisankarhsNo ratings yet
- KB FileDocument18 pagesKB FileKaushik BanikNo ratings yet
- Chapter Summary: Chapter 4: The Structure and Function of GenesDocument26 pagesChapter Summary: Chapter 4: The Structure and Function of GenesmongguNo ratings yet
- Growth Phase DependentDocument7 pagesGrowth Phase DependentzaraNo ratings yet
- New Yeast-Escicerlsa Shuttle Vectors Constructed With Iu Vitro Mutagenized Yeast Genes Lacking Six-Base Pair Restriction SitesDocument8 pagesNew Yeast-Escicerlsa Shuttle Vectors Constructed With Iu Vitro Mutagenized Yeast Genes Lacking Six-Base Pair Restriction SitesVijendra KavatalkarNo ratings yet
- Jurnal RFLP PDFDocument8 pagesJurnal RFLP PDFSilvia ZahiraNo ratings yet
- Gobatto 2014Document10 pagesGobatto 2014Francisca Beltrán GuzmánNo ratings yet
- Grapevine Viroids in ChileDocument2 pagesGrapevine Viroids in ChileFrancisca Beltrán GuzmánNo ratings yet
- Figueroa Et Al. - 2010 - Confirmation of The Presence of Citrus Viroids in ArgentinaDocument4 pagesFigueroa Et Al. - 2010 - Confirmation of The Presence of Citrus Viroids in ArgentinaFrancisca Beltrán GuzmánNo ratings yet
- Kitajima 2020 BrasilDocument101 pagesKitajima 2020 BrasilFrancisca Beltrán GuzmánNo ratings yet
- Herranz Et Al., 2013Document15 pagesHerranz Et Al., 2013Francisca Beltrán GuzmánNo ratings yet
- Sneath 1956Document29 pagesSneath 1956Francisca Beltrán GuzmánNo ratings yet
- ICVF 2015 APPLE N.FioreDocument1 pageICVF 2015 APPLE N.FioreFrancisca Beltrán GuzmánNo ratings yet
- Sweet Cherry 1973Document98 pagesSweet Cherry 1973Francisca Beltrán GuzmánNo ratings yet
- The Differentiation Basis Arginine Metabolism Other Gram-Negative Bacteria On TheDocument16 pagesThe Differentiation Basis Arginine Metabolism Other Gram-Negative Bacteria On TheFrancisca Beltrán GuzmánNo ratings yet
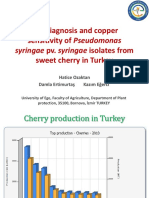
- The Diagnosis and Copper Sensitivity of Pseudomonas Sweet Cherry in TurkeyDocument33 pagesThe Diagnosis and Copper Sensitivity of Pseudomonas Sweet Cherry in TurkeyFrancisca Beltrán GuzmánNo ratings yet
- Genomic Investigation of Cherry Pathogenic Pseudomonas Syringae PathovarsDocument30 pagesGenomic Investigation of Cherry Pathogenic Pseudomonas Syringae PathovarsFrancisca Beltrán GuzmánNo ratings yet
- Otto2018 Article BacterialCankerOfCherryTreesPrDocument12 pagesOtto2018 Article BacterialCankerOfCherryTreesPrFrancisca Beltrán GuzmánNo ratings yet
- International Journal of Food Microbiology: Prashant Singh, Azlin MustaphaDocument8 pagesInternational Journal of Food Microbiology: Prashant Singh, Azlin MustaphaFrancisca Beltrán GuzmánNo ratings yet
- Kaluzna and Sobiczewski - 2009Document9 pagesKaluzna and Sobiczewski - 2009Francisca Beltrán GuzmánNo ratings yet
- Lebeis 2014Document7 pagesLebeis 2014Francisca Beltrán GuzmánNo ratings yet
- Wang 2006 VirulenceDocument14 pagesWang 2006 VirulenceFrancisca Beltrán GuzmánNo ratings yet
- Tsoktouridis Et Al 2014Document9 pagesTsoktouridis Et Al 2014Francisca Beltrán GuzmánNo ratings yet
- Bereswill 1994 PDFDocument7 pagesBereswill 1994 PDFFrancisca Beltrán GuzmánNo ratings yet
- Advances Sequencing Technologies - Methods and GoalsDocument10 pagesAdvances Sequencing Technologies - Methods and GoalsFrancisca Beltrán GuzmánNo ratings yet
- Comparison Techiques. 2012Document11 pagesComparison Techiques. 2012Francisca Beltrán GuzmánNo ratings yet
- Anti Egfr in MCRCDocument64 pagesAnti Egfr in MCRCHemanth KumarNo ratings yet
- GMOs - Environmental Concerns - Garden OrganicDocument1 pageGMOs - Environmental Concerns - Garden OrganicAndyPandyNo ratings yet
- Answer 1:: (Class - XII)Document6 pagesAnswer 1:: (Class - XII)Subhadip MurmuNo ratings yet
- Hepatoprotective Activity of Ocimum Americanum L LDocument7 pagesHepatoprotective Activity of Ocimum Americanum L LNurul Rachman NasutionNo ratings yet
- Review SheetDocument2 pagesReview Sheetapi-289866381No ratings yet
- Biotechnology Principles & ProcessesDocument3 pagesBiotechnology Principles & ProcessesKambaska Kumar BeheraNo ratings yet
- Lecture 3 - Bacterial Anatomy and PhysiologyDocument26 pagesLecture 3 - Bacterial Anatomy and Physiologyapi-370335280% (5)
- Inflammation and Types of Inflammation-1Document9 pagesInflammation and Types of Inflammation-1Shalabh JoharyNo ratings yet
- Digeorge Syndrome: Rupesh Mohandas GR3Document18 pagesDigeorge Syndrome: Rupesh Mohandas GR3Rupesh MohandasNo ratings yet
- Tobacco Etch Virus Protease: A Shortcut Across BiotechnologiesDocument16 pagesTobacco Etch Virus Protease: A Shortcut Across BiotechnologiesNhật ThiệnNo ratings yet
- Genetics of RetinoblastomaDocument5 pagesGenetics of RetinoblastomadesmawitaNo ratings yet
- AQA Immunity BookletDocument6 pagesAQA Immunity Bookletnchauhan212No ratings yet
- Heredity UnitDocument60 pagesHeredity Unitapi-224842598No ratings yet
- 7.2 Gaseous Exchange in PlantsDocument17 pages7.2 Gaseous Exchange in PlantsTheresa IzaNo ratings yet
- Stein 2010Document5 pagesStein 2010Eunice RamosNo ratings yet
- Difficulty Level Analysis NEETDocument4 pagesDifficulty Level Analysis NEETVinod Kumar JainNo ratings yet
- Abbreviated TitleDocument230 pagesAbbreviated TitleRico MamboNo ratings yet
- Genetic Disorders Associated With MeiosisDocument1 pageGenetic Disorders Associated With MeiosisJayrelle D. SafranNo ratings yet
- Taxonomy PDFDocument15 pagesTaxonomy PDFSwarup MondalNo ratings yet
- Yeast Cellular Respiration Labfin-2Document7 pagesYeast Cellular Respiration Labfin-2Nabeel UddinNo ratings yet
- 6.lymphatic SysDocument43 pages6.lymphatic SysMai Z HaniyaNo ratings yet
- Andrographis Paniculata A Review ofDocument12 pagesAndrographis Paniculata A Review oftio_bsNo ratings yet
- Perbedaan Dataran Tinggi Dan Dataran Rendah Terhadap Keberagaman Spesies Anopheles Spp. Di Provinsi Nusa Tenggara TimurDocument8 pagesPerbedaan Dataran Tinggi Dan Dataran Rendah Terhadap Keberagaman Spesies Anopheles Spp. Di Provinsi Nusa Tenggara TimurveryNo ratings yet
- Gene Expression and RegulationDocument27 pagesGene Expression and RegulationgengkapakNo ratings yet
- BiodiversityDocument94 pagesBiodiversitySaif Mohammad100% (1)
- The Evolution of PlantsDocument2 pagesThe Evolution of Plantsutopiayet7No ratings yet
- Course Catalogue 21 22 UpdatedDocument286 pagesCourse Catalogue 21 22 UpdatedKumarNo ratings yet
- Lab 6Document19 pagesLab 6nur athilahNo ratings yet
- Competency AddressedDocument46 pagesCompetency AddressedJaspreet SinghNo ratings yet
- Anatomy - Endocrine System - HormonesDocument21 pagesAnatomy - Endocrine System - HormonesShereen AlobinayNo ratings yet
- 10% Human: How Your Body's Microbes Hold the Key to Health and HappinessFrom Everand10% Human: How Your Body's Microbes Hold the Key to Health and HappinessRating: 4 out of 5 stars4/5 (33)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4.5 out of 5 stars4.5/5 (6)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisFrom EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisRating: 3.5 out of 5 stars3.5/5 (2)
- Tales from Both Sides of the Brain: A Life in NeuroscienceFrom EverandTales from Both Sides of the Brain: A Life in NeuroscienceRating: 3 out of 5 stars3/5 (18)
- The Rise and Fall of the Dinosaurs: A New History of a Lost WorldFrom EverandThe Rise and Fall of the Dinosaurs: A New History of a Lost WorldRating: 4 out of 5 stars4/5 (597)
- Buddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomFrom EverandBuddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomRating: 4 out of 5 stars4/5 (216)
- Return of the God Hypothesis: Three Scientific Discoveries That Reveal the Mind Behind the UniverseFrom EverandReturn of the God Hypothesis: Three Scientific Discoveries That Reveal the Mind Behind the UniverseRating: 4.5 out of 5 stars4.5/5 (52)
- A Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsFrom EverandA Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsRating: 4.5 out of 5 stars4.5/5 (6)
- The Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceFrom EverandThe Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceRating: 4.5 out of 5 stars4.5/5 (517)
- Gut: the new and revised Sunday Times bestsellerFrom EverandGut: the new and revised Sunday Times bestsellerRating: 4 out of 5 stars4/5 (393)
- Seven and a Half Lessons About the BrainFrom EverandSeven and a Half Lessons About the BrainRating: 4 out of 5 stars4/5 (111)
- Undeniable: How Biology Confirms Our Intuition That Life Is DesignedFrom EverandUndeniable: How Biology Confirms Our Intuition That Life Is DesignedRating: 4 out of 5 stars4/5 (11)
- All That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesFrom EverandAll That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesRating: 4.5 out of 5 stars4.5/5 (397)
- The Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionFrom EverandThe Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionRating: 4 out of 5 stars4/5 (812)
- Who's in Charge?: Free Will and the Science of the BrainFrom EverandWho's in Charge?: Free Will and the Science of the BrainRating: 4 out of 5 stars4/5 (65)
- Good Without God: What a Billion Nonreligious People Do BelieveFrom EverandGood Without God: What a Billion Nonreligious People Do BelieveRating: 4 out of 5 stars4/5 (66)
- Moral Tribes: Emotion, Reason, and the Gap Between Us and ThemFrom EverandMoral Tribes: Emotion, Reason, and the Gap Between Us and ThemRating: 4.5 out of 5 stars4.5/5 (116)
- Remnants of Ancient Life: The New Science of Old FossilsFrom EverandRemnants of Ancient Life: The New Science of Old FossilsRating: 3 out of 5 stars3/5 (3)
- The Lives of Bees: The Untold Story of the Honey Bee in the WildFrom EverandThe Lives of Bees: The Untold Story of the Honey Bee in the WildRating: 4.5 out of 5 stars4.5/5 (44)
- Darwin's Doubt: The Explosive Origin of Animal Life and the Case for Intelligent DesignFrom EverandDarwin's Doubt: The Explosive Origin of Animal Life and the Case for Intelligent DesignRating: 4 out of 5 stars4/5 (19)
- The Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindFrom EverandThe Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindRating: 4.5 out of 5 stars4.5/5 (93)
- Human: The Science Behind What Makes Your Brain UniqueFrom EverandHuman: The Science Behind What Makes Your Brain UniqueRating: 3.5 out of 5 stars3.5/5 (38)
- The Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorFrom EverandThe Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorNo ratings yet