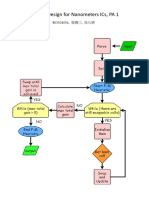
You might also like
- Static Security Assessment of Power System Using Kohonen Neural Network A. El-SharkawiDocument5 pagesStatic Security Assessment of Power System Using Kohonen Neural Network A. El-Sharkawirokhgireh_hojjatNo ratings yet
- PS2Document4 pagesPS2Romil ShahNo ratings yet
- PoS Forging Algorithms: Multi-Strategy Forging and Related Security IssuesDocument10 pagesPoS Forging Algorithms: Multi-Strategy Forging and Related Security IssuesandruimanNo ratings yet
- ReportDocument5 pagesReportNgô Trọng NguyênNo ratings yet
- Classification of Iris Data Set: Mentor: Assist. Prof. Primož Potočnik Student: Vitaly BorovinskiyDocument21 pagesClassification of Iris Data Set: Mentor: Assist. Prof. Primož Potočnik Student: Vitaly BorovinskiyrajendraNo ratings yet
- A Coordinated Multi-Switch Attack For Cascading Failures in Smart GridDocument12 pagesA Coordinated Multi-Switch Attack For Cascading Failures in Smart GridBo ChenNo ratings yet
- EE142 Project ReportDocument4 pagesEE142 Project Reportdi55olve88No ratings yet
- Classification of Iris Data SetDocument16 pagesClassification of Iris Data SetSofiene GuedriNo ratings yet
- Classification of Iris Data Set PDFDocument21 pagesClassification of Iris Data Set PDFSofiene GuedriNo ratings yet
- Matlab Iris RBFDocument21 pagesMatlab Iris RBFЯу МдNo ratings yet
- SisDocument21 pagesSistemp759No ratings yet
- Apply Genetic Algorithm To The Learning Phase of A Neural NetworkDocument6 pagesApply Genetic Algorithm To The Learning Phase of A Neural Networkjkl316No ratings yet
- Fuzzy ART Neural Network Algorithm For Classifying The Power System FaultsDocument9 pagesFuzzy ART Neural Network Algorithm For Classifying The Power System FaultsSwathy SasidharanNo ratings yet
- Variable Importance in NeuralDocument5 pagesVariable Importance in NeuralSharathKumarNo ratings yet
- Analysing Time Course Microarray Data Using Bioconductor: A Case Study Using Yeast2 Affymetrix ArraysDocument10 pagesAnalysing Time Course Microarray Data Using Bioconductor: A Case Study Using Yeast2 Affymetrix ArraysPavan KumarNo ratings yet
- W-Cdma Capacity Analysis Using Gis Based Planing Tools and Matlab SimulationDocument5 pagesW-Cdma Capacity Analysis Using Gis Based Planing Tools and Matlab SimulationanavarrocadavidNo ratings yet
- Mexican Hat NetworkDocument10 pagesMexican Hat NetworkSushant Ranade33% (3)
- Neural Network Weight Selection Using Genetic AlgorithmsDocument17 pagesNeural Network Weight Selection Using Genetic AlgorithmsChevalier De BalibariNo ratings yet
- Eiaicc02 035 3783233Document14 pagesEiaicc02 035 3783233marvel homesNo ratings yet
- NIHMS1845306-supplement-1845306 Sup MaterialsDocument22 pagesNIHMS1845306-supplement-1845306 Sup MaterialsCristian VillarNo ratings yet
- Ms - A Program For Generating Samples Under Neutral Models: Richard R. Hudson October 16, 2017Document22 pagesMs - A Program For Generating Samples Under Neutral Models: Richard R. Hudson October 16, 2017birdguy.saikat9936No ratings yet
- ZzzChapter 3 - Section 3.3.8Document11 pagesZzzChapter 3 - Section 3.3.8Buddhi ChinthakaNo ratings yet
- Data GenDocument4 pagesData GenAbeda KarimNo ratings yet
- Homework 1Document2 pagesHomework 1Hendra LimNo ratings yet
- Neural Network EnsemblesDocument9 pagesNeural Network EnsembleskronalizedNo ratings yet
- ChapterThree NeuralNetworksDocument10 pagesChapterThree NeuralNetworksBO-IB MICHAELNo ratings yet
- 0491 ThB15 6Document6 pages0491 ThB15 6Peerapol YuvapoositanonNo ratings yet
- Experiment 11 - DNA: Biomedical Computation Lab (2) :: Ngee Ann Polytechnic Medical Imaging, Informatics and TelemedicineDocument5 pagesExperiment 11 - DNA: Biomedical Computation Lab (2) :: Ngee Ann Polytechnic Medical Imaging, Informatics and TelemedicinedavidwongbxNo ratings yet
- Complex Network ReportDocument16 pagesComplex Network ReportMohit SharmaNo ratings yet
- Active circuitsGADocument6 pagesActive circuitsGASn ProfNo ratings yet
- Overview and Tutorial On Artificial Intelligence Systems: Eric ConradDocument6 pagesOverview and Tutorial On Artificial Intelligence Systems: Eric ConradEhab SabryNo ratings yet
- Using Random Forests v4.0Document33 pagesUsing Random Forests v4.0rollschachNo ratings yet
- Vasilic2005Document9 pagesVasilic2005fernando ceballosNo ratings yet
- Final Project Report: Sequential Circuit Simulator With FaultsDocument10 pagesFinal Project Report: Sequential Circuit Simulator With FaultszeSky ArmourNo ratings yet
- Cellular Automata in Public Key CryptographyDocument8 pagesCellular Automata in Public Key CryptographyIJERAS-International Journal of Engineering Research and Applied Science (ISSN: 2349-4522)No ratings yet
- Experiment No 4Document3 pagesExperiment No 421bme145No ratings yet
- Approximation of Large-Scale Dynamical Systems: An Overview: A.C. Antoulas and D.C. Sorensen August 31, 2001Document22 pagesApproximation of Large-Scale Dynamical Systems: An Overview: A.C. Antoulas and D.C. Sorensen August 31, 2001Anonymous lEBdswQXmxNo ratings yet
- Paper1 PDFDocument12 pagesPaper1 PDFBo ChenNo ratings yet
- Cell-Free Massive MIMO SystemsDocument5 pagesCell-Free Massive MIMO SystemsMalakNo ratings yet
- Interview DVDocument5 pagesInterview DVPrasad Reddy100% (1)
- End Sem PresentationDocument4 pagesEnd Sem PresentationShady dtNo ratings yet
- Conglomerate Inc.'s New PDA: A Segmentation Study: Case HandoutDocument13 pagesConglomerate Inc.'s New PDA: A Segmentation Study: Case HandoutTinaPapadopoulouNo ratings yet
- 9bus System, Power FlowDocument17 pages9bus System, Power FlowZeeshan AhmedNo ratings yet
- Machine Learning AssignmentDocument8 pagesMachine Learning AssignmentJoshuaDownesNo ratings yet
- F09 E6 CB0 D 01Document10 pagesF09 E6 CB0 D 01Laxmansarat KondepudiNo ratings yet
- Agilent TXT File Pre-Processing Engine PDFDocument42 pagesAgilent TXT File Pre-Processing Engine PDFscjofyWFawlroa2r06YFVabfbajNo ratings yet
- 6 Genetic Algorithms: 6.1 TheoryDocument30 pages6 Genetic Algorithms: 6.1 TheoryRodolfo FloresNo ratings yet
- Real Timepower System Security - EEEDocument7 pagesReal Timepower System Security - EEEpradeep9007879No ratings yet
- International Journal of Computer Science: Theory and ApplicationDocument6 pagesInternational Journal of Computer Science: Theory and ApplicationijcstaNo ratings yet
- Institut de Ciencia de Materials de Barcelona, CSIC Camus de La UAB, 08193 Barcelona, SpainDocument8 pagesInstitut de Ciencia de Materials de Barcelona, CSIC Camus de La UAB, 08193 Barcelona, SpainManjeet BhatiaNo ratings yet
- Pattern RecongnitionDocument21 pagesPattern RecongnitionRiad El AbedNo ratings yet
- Towards Scalable and Robust Overlay Networks: Baruch Awerbuch Christian ScheidelerDocument6 pagesTowards Scalable and Robust Overlay Networks: Baruch Awerbuch Christian ScheidelerNghĩa ZerNo ratings yet
- CORPOELECDocument33 pagesCORPOELECjoelNo ratings yet
- C22 NRSC2001 Dynamic Generation of S-Boxes in Block Cipher SystemsDocument9 pagesC22 NRSC2001 Dynamic Generation of S-Boxes in Block Cipher SystemsDr.Hesham El-BadawyNo ratings yet
- WPO 10 WCDMA Neighbor Cell Scrambling Code and LAC Planning 47Document47 pagesWPO 10 WCDMA Neighbor Cell Scrambling Code and LAC Planning 47Maxime de SouzaNo ratings yet
- Learning Vector Quantization (LVQ) and K-Nearest Neighbor For Intrusion ClassificationDocument5 pagesLearning Vector Quantization (LVQ) and K-Nearest Neighbor For Intrusion ClassificationWorld of Computer Science and Information Technology JournalNo ratings yet
- SV Interview BookDocument11 pagesSV Interview Bookmaheshdesh1617No ratings yet
- Time Domain Structural Health Monitoring With Magnetostrictive Patches Using Five Stage Hierarchical Neural NetworksDocument7 pagesTime Domain Structural Health Monitoring With Magnetostrictive Patches Using Five Stage Hierarchical Neural NetworksDebiprasad GhoshNo ratings yet
- CS395T - Computational Statistics With Application To BioinformaticsDocument5 pagesCS395T - Computational Statistics With Application To Bioinformaticspanna1No ratings yet
- Current Topics in BioinformaticsDocument21 pagesCurrent Topics in Bioinformaticspanna1No ratings yet
- Symmetric Matrices and Orthogonal DiagonalizationDocument15 pagesSymmetric Matrices and Orthogonal Diagonalizationpanna1No ratings yet
- Systems Biology BNF079: Preliminary Schedule Fall 2004Document1 pageSystems Biology BNF079: Preliminary Schedule Fall 2004panna1No ratings yet
- Name Synopsis Description: Lefty User GuideDocument1 pageName Synopsis Description: Lefty User Guidepanna1No ratings yet
- Print Name:: EECS 206, Winter 2002 FINAL EXAM Mond. Apr. 22, 2002 - 4:00-6:00pmDocument1 pagePrint Name:: EECS 206, Winter 2002 FINAL EXAM Mond. Apr. 22, 2002 - 4:00-6:00pmpanna1No ratings yet
- Heat MapDocument1 pageHeat Mappanna1No ratings yet
- Name Synopsis: X + 2 Edges If FoldedDocument1 pageName Synopsis: X + 2 Edges If Foldedpanna1No ratings yet
- gxl2gv 1 PDFDocument1 pagegxl2gv 1 PDFpanna1No ratings yet
- The Role of Knowledge Management and Internal CommDocument9 pagesThe Role of Knowledge Management and Internal CommsamNo ratings yet
- 7th MEXICAN CULTUREDocument3 pages7th MEXICAN CULTUREJosé Carlos Villatoro AlvaradoNo ratings yet
- Electrical Electronic Devices ShabbatDocument79 pagesElectrical Electronic Devices Shabbatמאירה הדרNo ratings yet
- Past Simple Regular Verbs Fun Activities Games - 18699Document2 pagesPast Simple Regular Verbs Fun Activities Games - 18699emilyNo ratings yet
- The Impact of Culture On The Quality of Internal Audit An Empirical Study.Document22 pagesThe Impact of Culture On The Quality of Internal Audit An Empirical Study.Vaisal AmirNo ratings yet
- Negotiation and Diplomacy Term PaperDocument37 pagesNegotiation and Diplomacy Term PaperManifa OsmanNo ratings yet
- Cheiloscopy in Gender Determination: A Study On 2112 IndividualsDocument5 pagesCheiloscopy in Gender Determination: A Study On 2112 Individualsknr crsNo ratings yet
- GPS Chapter 05. Binding TheoryDocument5 pagesGPS Chapter 05. Binding TheoryAhsan Raza100% (5)
- FileContent 9Document103 pagesFileContent 9Samadashvili ZukaNo ratings yet
- Full Download Introduction To Operations and Supply Chain Management 3rd Edition Bozarth Test BankDocument36 pagesFull Download Introduction To Operations and Supply Chain Management 3rd Edition Bozarth Test Bankcoraleementgen1858100% (42)
- ADL Audit of Anti-Semitic Incidents 2021Document38 pagesADL Audit of Anti-Semitic Incidents 2021Michael_Roberts2019No ratings yet
- Haunted Castles IrelandDocument3 pagesHaunted Castles IrelandRenata TatomirNo ratings yet
- Chemistry MCQs Test HandoutsDocument28 pagesChemistry MCQs Test HandoutsOsama Hasan88% (24)
- The Birth of The Bipolar Disorder: Historical ReviewDocument10 pagesThe Birth of The Bipolar Disorder: Historical ReviewFELIPE ABREONo ratings yet
- Answer Key: Cumulative TestDocument2 pagesAnswer Key: Cumulative TestTeodora MitrovicNo ratings yet
- Cardiohelp System Brochure-En-Non Us JapanDocument16 pagesCardiohelp System Brochure-En-Non Us JapanAnar MaharramovNo ratings yet
- Week 3 CPAR Day 1Document4 pagesWeek 3 CPAR Day 1zessicrel mejiasNo ratings yet
- Future 100 2019Document237 pagesFuture 100 2019Maruam Samek100% (1)
- 15 UtilitarianismDocument25 pages15 UtilitarianismGracelyn CablayNo ratings yet
- Human Medicinal Agents From PlantsDocument358 pagesHuman Medicinal Agents From Plantsamino12451100% (1)
- Team Building Breakout ScriptDocument12 pagesTeam Building Breakout ScriptMelanie PajaronNo ratings yet
- Jeremy Bentham - A Quick SummaryDocument2 pagesJeremy Bentham - A Quick SummaryDebashish DashNo ratings yet
- Japanese Horror Films and Their American Remakes 9780203382448 - WebpdfDocument273 pagesJapanese Horror Films and Their American Remakes 9780203382448 - WebpdfAmbrose66No ratings yet
- Examples of FallaciesDocument2 pagesExamples of FallaciesSinta HandayaniNo ratings yet
- Critical Analysis of Sonnet 18Document9 pagesCritical Analysis of Sonnet 18Shaina Eleccion50% (4)
- FCTM Normal Emergency & Abnormal Procedures (Pec Version) m514v1 PDFDocument170 pagesFCTM Normal Emergency & Abnormal Procedures (Pec Version) m514v1 PDFcapt.sahaag100% (14)
- The Authentic I Ching The Three Classic Methods of PredictionDocument4 pagesThe Authentic I Ching The Three Classic Methods of Predictionmaximo0% (1)
- Daftar Nama Dosen Pembimbing Periode 2020-1 PagiDocument3 pagesDaftar Nama Dosen Pembimbing Periode 2020-1 PagiNufita Twandita DewiNo ratings yet
- EDS261Document8 pagesEDS261micasandogu01No ratings yet
- English Case: Davis Contractors LTD V Fareham UDCDocument7 pagesEnglish Case: Davis Contractors LTD V Fareham UDCAin AnuwarNo ratings yet
- Four Battlegrounds: Power in the Age of Artificial IntelligenceFrom EverandFour Battlegrounds: Power in the Age of Artificial IntelligenceRating: 5 out of 5 stars5/5 (5)
- ChatGPT Side Hustles 2024 - Unlock the Digital Goldmine and Get AI Working for You Fast with More Than 85 Side Hustle Ideas to Boost Passive Income, Create New Cash Flow, and Get Ahead of the CurveFrom EverandChatGPT Side Hustles 2024 - Unlock the Digital Goldmine and Get AI Working for You Fast with More Than 85 Side Hustle Ideas to Boost Passive Income, Create New Cash Flow, and Get Ahead of the CurveNo ratings yet
- Generative AI: The Insights You Need from Harvard Business ReviewFrom EverandGenerative AI: The Insights You Need from Harvard Business ReviewRating: 4.5 out of 5 stars4.5/5 (2)
- Scary Smart: The Future of Artificial Intelligence and How You Can Save Our WorldFrom EverandScary Smart: The Future of Artificial Intelligence and How You Can Save Our WorldRating: 4.5 out of 5 stars4.5/5 (55)
- The Master Algorithm: How the Quest for the Ultimate Learning Machine Will Remake Our WorldFrom EverandThe Master Algorithm: How the Quest for the Ultimate Learning Machine Will Remake Our WorldRating: 4.5 out of 5 stars4.5/5 (107)
- 100M Offers Made Easy: Create Your Own Irresistible Offers by Turning ChatGPT into Alex HormoziFrom Everand100M Offers Made Easy: Create Your Own Irresistible Offers by Turning ChatGPT into Alex HormoziNo ratings yet
- ChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindFrom EverandChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindNo ratings yet
- HBR's 10 Must Reads on AI, Analytics, and the New Machine AgeFrom EverandHBR's 10 Must Reads on AI, Analytics, and the New Machine AgeRating: 4.5 out of 5 stars4.5/5 (69)
- Working with AI: Real Stories of Human-Machine Collaboration (Management on the Cutting Edge)From EverandWorking with AI: Real Stories of Human-Machine Collaboration (Management on the Cutting Edge)Rating: 5 out of 5 stars5/5 (5)
- The AI Advantage: How to Put the Artificial Intelligence Revolution to WorkFrom EverandThe AI Advantage: How to Put the Artificial Intelligence Revolution to WorkRating: 4 out of 5 stars4/5 (7)
- Who's Afraid of AI?: Fear and Promise in the Age of Thinking MachinesFrom EverandWho's Afraid of AI?: Fear and Promise in the Age of Thinking MachinesRating: 4.5 out of 5 stars4.5/5 (13)
- Artificial Intelligence: A Guide for Thinking HumansFrom EverandArtificial Intelligence: A Guide for Thinking HumansRating: 4.5 out of 5 stars4.5/5 (30)
- System Design Interview: 300 Questions And Answers: Prepare And PassFrom EverandSystem Design Interview: 300 Questions And Answers: Prepare And PassNo ratings yet
- Rise of Generative AI and ChatGPT: Understand how Generative AI and ChatGPT are transforming and reshaping the business world (English Edition)From EverandRise of Generative AI and ChatGPT: Understand how Generative AI and ChatGPT are transforming and reshaping the business world (English Edition)No ratings yet
- Artificial Intelligence & Generative AI for Beginners: The Complete GuideFrom EverandArtificial Intelligence & Generative AI for Beginners: The Complete GuideRating: 5 out of 5 stars5/5 (1)
- The Digital Mind: How Science is Redefining HumanityFrom EverandThe Digital Mind: How Science is Redefining HumanityRating: 4.5 out of 5 stars4.5/5 (2)
- Python Machine Learning - Third Edition: Machine Learning and Deep Learning with Python, scikit-learn, and TensorFlow 2, 3rd EditionFrom EverandPython Machine Learning - Third Edition: Machine Learning and Deep Learning with Python, scikit-learn, and TensorFlow 2, 3rd EditionRating: 5 out of 5 stars5/5 (2)
- How to Make Money Online Using ChatGPT Prompts: Secrets Revealed for Unlocking Hidden Opportunities. Earn Full-Time Income Using ChatGPT with the Untold Potential of Conversational AI.From EverandHow to Make Money Online Using ChatGPT Prompts: Secrets Revealed for Unlocking Hidden Opportunities. Earn Full-Time Income Using ChatGPT with the Untold Potential of Conversational AI.No ratings yet
- ChatGPT Millionaire 2024 - Bot-Driven Side Hustles, Prompt Engineering Shortcut Secrets, and Automated Income Streams that Print Money While You Sleep. The Ultimate Beginner’s Guide for AI BusinessFrom EverandChatGPT Millionaire 2024 - Bot-Driven Side Hustles, Prompt Engineering Shortcut Secrets, and Automated Income Streams that Print Money While You Sleep. The Ultimate Beginner’s Guide for AI BusinessNo ratings yet
- Architects of Intelligence: The truth about AI from the people building itFrom EverandArchitects of Intelligence: The truth about AI from the people building itRating: 4.5 out of 5 stars4.5/5 (21)
- Artificial Intelligence: The Insights You Need from Harvard Business ReviewFrom EverandArtificial Intelligence: The Insights You Need from Harvard Business ReviewRating: 4.5 out of 5 stars4.5/5 (104)
- T-Minus AI: Humanity's Countdown to Artificial Intelligence and the New Pursuit of Global PowerFrom EverandT-Minus AI: Humanity's Countdown to Artificial Intelligence and the New Pursuit of Global PowerRating: 4 out of 5 stars4/5 (4)
- Make Money with ChatGPT: Your Guide to Making Passive Income Online with Ease using AI: AI Wealth MasteryFrom EverandMake Money with ChatGPT: Your Guide to Making Passive Income Online with Ease using AI: AI Wealth MasteryNo ratings yet