You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5807)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Chapter 10 Haloalkanes and HaloarenesDocument24 pagesChapter 10 Haloalkanes and HaloarenesSuhas GowdaNo ratings yet
- Alkene Reaction GuideDocument41 pagesAlkene Reaction GuideAbhishek Isaac MathewNo ratings yet
- Chemistry Syllabus (BS)Document111 pagesChemistry Syllabus (BS)Abdur Rehman80% (5)
- Chemistry Notes Class 11 Chapter 13 HydrocarbonsDocument26 pagesChemistry Notes Class 11 Chapter 13 HydrocarbonsShashank PratapNo ratings yet
- Beginning Organic Chemistry 2Document2 pagesBeginning Organic Chemistry 2Daniel Alejandro Rojas ToroNo ratings yet
- Simulation of Chlorobenzene Plant by Using Aspen PlusDocument10 pagesSimulation of Chlorobenzene Plant by Using Aspen PlusamalNo ratings yet
- An Advanced Inorganic Chemistry Experiment: Synthesis and Symmetry of Two Cobalt (III) Complexes With Tetradentate LigandsDocument2 pagesAn Advanced Inorganic Chemistry Experiment: Synthesis and Symmetry of Two Cobalt (III) Complexes With Tetradentate LigandsFausto SalazarNo ratings yet
- Bis (Dimethylglyoximat0) Cobalt: ReferencesDocument10 pagesBis (Dimethylglyoximat0) Cobalt: ReferencesFausto SalazarNo ratings yet
- Tautomerism of 1,2,3 - and 1,2,4-Triazole in The Gas Phase and in Aqueous Solution - JPC 1990Document3 pagesTautomerism of 1,2,3 - and 1,2,4-Triazole in The Gas Phase and in Aqueous Solution - JPC 1990Fausto SalazarNo ratings yet
- Ring-Chain Tautomerism - Valters, Flitsch, 1985 PDFDocument284 pagesRing-Chain Tautomerism - Valters, Flitsch, 1985 PDFFausto SalazarNo ratings yet
- Chapter 9 Questions and AnswersDocument8 pagesChapter 9 Questions and AnswersFausto SalazarNo ratings yet
- Helvetica Chimica ActaDocument1 pageHelvetica Chimica ActaFausto SalazarNo ratings yet
- Some Observations On Molecular Orbital Theory PDFDocument5 pagesSome Observations On Molecular Orbital Theory PDFFausto SalazarNo ratings yet
- Lindquist1997 PDFDocument2 pagesLindquist1997 PDFFausto SalazarNo ratings yet
- Blatter 1990Document3 pagesBlatter 1990Fausto SalazarNo ratings yet
- Ionization Energies of Atoms and Atomic Ions PDFDocument9 pagesIonization Energies of Atoms and Atomic Ions PDFFausto SalazarNo ratings yet
- Gravimetric Determination of Calcium As CaC2O4 - H2ODocument1 pageGravimetric Determination of Calcium As CaC2O4 - H2OFausto SalazarNo ratings yet
- Introducing Copper As Catalyst For Oxidative Alkane DehydrogenationDocument10 pagesIntroducing Copper As Catalyst For Oxidative Alkane DehydrogenationFausto SalazarNo ratings yet
- Video Questions Related To The Case of The Missing HareDocument2 pagesVideo Questions Related To The Case of The Missing HareFausto SalazarNo ratings yet
- Formal Report 1Document6 pagesFormal Report 1Patricia Denise OrquiaNo ratings yet
- Reasoning Ques in Organic ChemistryDocument14 pagesReasoning Ques in Organic ChemistryRIHINBHATNAGAR50% (2)
- Chem 31 (Upm)Document7 pagesChem 31 (Upm)Patricia Gayle JacildoNo ratings yet
- Pre-Medical: Chemistry Allen: Carbonyl Compounds, Acids and It'S Derivatives Carbonyl CompoundsDocument18 pagesPre-Medical: Chemistry Allen: Carbonyl Compounds, Acids and It'S Derivatives Carbonyl CompoundsJK JHANo ratings yet
- NEET Chemistry SyllabusDocument10 pagesNEET Chemistry SyllabusRiyaz ShaikhNo ratings yet
- M.Sc. Chemistry 9 - 3 - 17Document87 pagesM.Sc. Chemistry 9 - 3 - 17rahulNo ratings yet
- Chemistry PPT 21508Document1 pageChemistry PPT 21508Sunil SharmaNo ratings yet
- Alkyl Halides & Aryl Halides: Victor GrignardDocument50 pagesAlkyl Halides & Aryl Halides: Victor GrignardsarahNo ratings yet
- Tetrahydro Quino LinesDocument40 pagesTetrahydro Quino LinesРумен ЛяпчевNo ratings yet
- 10.4 Hydroxy Compounds Alcohols: Learning OutcomesDocument10 pages10.4 Hydroxy Compounds Alcohols: Learning OutcomesSaqib HussainNo ratings yet
- Senyawa KarbonilDocument61 pagesSenyawa KarbonilsalmaNo ratings yet
- IJC H2 Paper 1 and 2 Answers (For Sharing)Document9 pagesIJC H2 Paper 1 and 2 Answers (For Sharing)Sharon HowNo ratings yet
- Bromine Water Test For PhenolsDocument6 pagesBromine Water Test For Phenolscleahis cruzNo ratings yet
- Chapter 11 SolutionsDocument24 pagesChapter 11 SolutionsChelley Sharleene PecajasNo ratings yet
- 2nd Chem Shortlisting-1Document20 pages2nd Chem Shortlisting-1Mudassar AbbasNo ratings yet
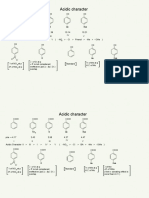
- Acidic Character of Phenols CarboxilicAcid AKSAMALDocument15 pagesAcidic Character of Phenols CarboxilicAcid AKSAMALsantoshguptaaNo ratings yet
- III) GOC-Some Basic Principles & Techniques: 1. Priority Order F.G. Multiple Bond SubstituentDocument15 pagesIII) GOC-Some Basic Principles & Techniques: 1. Priority Order F.G. Multiple Bond SubstituentSHIVAMNo ratings yet
- Physical and Chemical Properties of AlcoholsDocument24 pagesPhysical and Chemical Properties of AlcoholsmeerasahibfarhanNo ratings yet
- Halogen CompoundsDocument43 pagesHalogen CompoundsSai Sasivardhan GampaNo ratings yet
- Class 12 Alcohols Phenols Ethers NotesDocument23 pagesClass 12 Alcohols Phenols Ethers NotesIpsita SethiNo ratings yet
- 5-EAS Spring 14Document10 pages5-EAS Spring 14Prashanth CiryamNo ratings yet
- Effect of Substituents On ReactivityDocument7 pagesEffect of Substituents On ReactivityWebster KafungaNo ratings yet
- 01 Chemistry Teaching & Test Schedules@28!06!21 5.00PmDocument5 pages01 Chemistry Teaching & Test Schedules@28!06!21 5.00PmStaus world IndiaNo ratings yet
- Kimia Organik: Prof. Setiyo Gunawan, ST., Ph.D. Chemical Engineering Department Fti-ItsDocument7 pagesKimia Organik: Prof. Setiyo Gunawan, ST., Ph.D. Chemical Engineering Department Fti-ItsMohammad Farhan SNo ratings yet