You might also like
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- Physiologic Monitoring of The Surgical PatientDocument56 pagesPhysiologic Monitoring of The Surgical PatientSeid Adem100% (2)
- Ug NotesDocument538 pagesUg NotesSteven IStudy SmithNo ratings yet
- Nclex Random Fact To StudyDocument431 pagesNclex Random Fact To StudyNeko Neko100% (1)
- Pediatric Antibiotic Dosing Card 2012Document2 pagesPediatric Antibiotic Dosing Card 2012yoshilimsiacoshigyoNo ratings yet
- Sankara Nethralaya's Manual of Medical Laboratory TechniquesDocument453 pagesSankara Nethralaya's Manual of Medical Laboratory Techniquesmahmoudbadr291% (45)
- Ah4379200077182260 RLSDocument10 pagesAh4379200077182260 RLSAnusha NNo ratings yet
- Typical Limit of DEFLECTIONDocument4 pagesTypical Limit of DEFLECTIONfreddielistoNo ratings yet
- Acute Kidney Injury W/ Hyperkalemia NCPDocument5 pagesAcute Kidney Injury W/ Hyperkalemia NCPMyrvic Ortiz La OrdenNo ratings yet
- Effect of Hepatic and Renal Impairment On PharmacokineticDocument46 pagesEffect of Hepatic and Renal Impairment On PharmacokineticPURVI NEEMANo ratings yet
- Consolidation ReportDocument9 pagesConsolidation ReportKhaled RamadanNo ratings yet
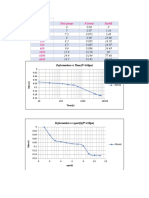
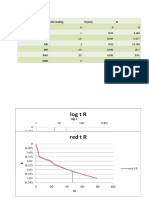
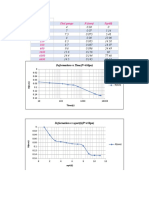
- Time (Sec) Dial Gauge R (MM) SQRT (T) 0 10 30 120 210 360 600 3600 6000 6600Document1 pageTime (Sec) Dial Gauge R (MM) SQRT (T) 0 10 30 120 210 360 600 3600 6000 6600Khaled RamadanNo ratings yet
- P 20 Kpa: Time (Sec) Dial Reading R (MM) TDocument1 pageP 20 Kpa: Time (Sec) Dial Reading R (MM) TKhaled RamadanNo ratings yet
- Time (Sec) Dial Gauge R (MM) SQRT (T) 0 10 30 120 210 360 600 3600 6000 6600Document1 pageTime (Sec) Dial Gauge R (MM) SQRT (T) 0 10 30 120 210 360 600 3600 6000 6600Khaled RamadanNo ratings yet
- Time (Sec) Dial Gauge R (MM) SQRT (T) 0 10 30 120 210 360 600 3600 6000 6600Document1 pageTime (Sec) Dial Gauge R (MM) SQRT (T) 0 10 30 120 210 360 600 3600 6000 6600Khaled RamadanNo ratings yet
- Consolidation ReportDocument9 pagesConsolidation ReportKhaled RamadanNo ratings yet
- BIM - Assignment 1: Prof. Yehya Temsah & Dr. Ali JahamiDocument6 pagesBIM - Assignment 1: Prof. Yehya Temsah & Dr. Ali JahamiKhaled RamadanNo ratings yet
- 2020 SAFAR - Call For Applications 6.2.2020 Final Withour RedDocument3 pages2020 SAFAR - Call For Applications 6.2.2020 Final Withour RedKhaled RamadanNo ratings yet
- Seismic Retrofit of RC Frame Buildings With Masonry Infill Walls: Literature Review and Preliminary Case StudyDocument83 pagesSeismic Retrofit of RC Frame Buildings With Masonry Infill Walls: Literature Review and Preliminary Case Studybasabi12No ratings yet
- NSPE Code of Ethics For EngineersDocument2 pagesNSPE Code of Ethics For EngineersYob YnnosNo ratings yet
- Difference in Floors Loading DistributionDocument2 pagesDifference in Floors Loading DistributionKhaled RamadanNo ratings yet
- Book Lecture PDFDocument118 pagesBook Lecture PDFKhaled RamadanNo ratings yet
- Super Imposed Dead Load (SIDL) :: Quality X Density KN M KN M KN M KN M Total 0.46+0.66+0.96+ 0.44 2.5 KN MDocument15 pagesSuper Imposed Dead Load (SIDL) :: Quality X Density KN M KN M KN M KN M Total 0.46+0.66+0.96+ 0.44 2.5 KN MKhaled RamadanNo ratings yet
- MATH 283 - Part 3 - (Spring 14-15) PDFDocument7 pagesMATH 283 - Part 3 - (Spring 14-15) PDFShawki BsatNo ratings yet
- Fourier Series Is A Trigonometric Series of The Form: A NX NX FX A Cos B P PDocument8 pagesFourier Series Is A Trigonometric Series of The Form: A NX NX FX A Cos B P PKhaled RamadanNo ratings yet
- Objectivity and Truthfulness-Use of Drone Case No. 18-11 FactsDocument4 pagesObjectivity and Truthfulness-Use of Drone Case No. 18-11 FactsKhaled RamadanNo ratings yet
- FX X P: Half-Range ExpansionDocument9 pagesFX X P: Half-Range ExpansionKhaled RamadanNo ratings yet
- Dy Pxy Fxy DX: Bernoulli's EquationDocument6 pagesDy Pxy Fxy DX: Bernoulli's Equationahmadomar89No ratings yet
- RC II Project Excel SheetDocument33 pagesRC II Project Excel SheetKhaled RamadanNo ratings yet
- MATH 283 - Part 1 - (Spring 14-15)Document19 pagesMATH 283 - Part 1 - (Spring 14-15)Shawki BsatNo ratings yet
- Lec 7Document4 pagesLec 7Khaled RamadanNo ratings yet
- HW 2 (L.T) PDFDocument1 pageHW 2 (L.T) PDFKhaled RamadanNo ratings yet
- A Power Series in (X-A) Is An Finite Series of The FormDocument6 pagesA Power Series in (X-A) Is An Finite Series of The FormKhaled RamadanNo ratings yet
- Free PPT TemplatesDocument4 pagesFree PPT TemplatesicaeeeNo ratings yet
- Nguyen T Et Al - 2017 - Favipiravir Phamacokinetics in Ebola Infected Patients of Jiki Trial PDFDocument19 pagesNguyen T Et Al - 2017 - Favipiravir Phamacokinetics in Ebola Infected Patients of Jiki Trial PDFKhaled RamadanNo ratings yet
- Antimicrobial Agents and Chemotherapy-2010-Sleeman-2517.fullDocument8 pagesAntimicrobial Agents and Chemotherapy-2010-Sleeman-2517.fullKhaled RamadanNo ratings yet
- Elevated Rectangular Water Tank DesignDocument12 pagesElevated Rectangular Water Tank DesignKhaled Ramadan0% (1)
- Nguyen T Et Al - 2017 - Favipiravir Phamacokinetics in Ebola Infected Patients of Jiki Trial PDFDocument19 pagesNguyen T Et Al - 2017 - Favipiravir Phamacokinetics in Ebola Infected Patients of Jiki Trial PDFKhaled RamadanNo ratings yet
- Report 856Document14 pagesReport 856Piyush RanjanNo ratings yet
- Toradol Product MonogrphDocument33 pagesToradol Product MonogrphRizka RamdhaniNo ratings yet
- 5Document10 pages5khalid alharbiNo ratings yet
- Creatinine RocheDocument2 pagesCreatinine Rocheadenos14No ratings yet
- Literature 11Document9 pagesLiterature 11Bayu SuyantoNo ratings yet
- Anemia in DiabetesDocument12 pagesAnemia in DiabetesRishitha MadhusudhanNo ratings yet
- Determining The Glomerular Filtration Rate: Andani Puspita Rani 1315244Document15 pagesDetermining The Glomerular Filtration Rate: Andani Puspita Rani 1315244AndreasJoviantoNo ratings yet
- Paraquat PoisoningDocument45 pagesParaquat PoisoningM. O. PHC HOLAVANAHALLYNo ratings yet
- Biochemical Tests in Diabetes: DR Joe Fleming PHD MCB Frcpath Dept of Clinical Biochemistry CMC VelloreDocument43 pagesBiochemical Tests in Diabetes: DR Joe Fleming PHD MCB Frcpath Dept of Clinical Biochemistry CMC VelloreTRINEL ANGGRIATINo ratings yet
- Unit 1 Pharmacology NotesDocument4 pagesUnit 1 Pharmacology Notessalted fishNo ratings yet
- VireadDocument17 pagesVireadastri rahayuNo ratings yet
- Sil4 TB 19Document29 pagesSil4 TB 19Zuriel AlmeydaNo ratings yet
- Hepatorrenal PrimerDocument17 pagesHepatorrenal PrimerDianaScandaOlveraCabreraNo ratings yet
- Approachtoelectrolyte Abnormalities, Prerenal Azotemia, AndfluidbalanceDocument15 pagesApproachtoelectrolyte Abnormalities, Prerenal Azotemia, AndfluidbalanceCecilia GrayebNo ratings yet
- Protokol KemoterapiDocument147 pagesProtokol KemoterapiDala VW100% (1)
- Dr. Poon's Metabolic Diet Program Improves Clinical and Laboratory Outcomes in Obese Patients With Metabolic SyndromeDocument5 pagesDr. Poon's Metabolic Diet Program Improves Clinical and Laboratory Outcomes in Obese Patients With Metabolic SyndromeasclepiuspdfsNo ratings yet
- Screenshot 2023-03-30 at 9.08.15 PMDocument14 pagesScreenshot 2023-03-30 at 9.08.15 PMUrvi KaliaNo ratings yet
- Clinical Biochemistry - Fifth YearDocument176 pagesClinical Biochemistry - Fifth YearBayan AlsaadiNo ratings yet
- Articoli Dante Trudel Integrazione (Articoli Facebook)Document11 pagesArticoli Dante Trudel Integrazione (Articoli Facebook)Matteo PanciroliNo ratings yet
- Reflotron Plus Information BookletDocument28 pagesReflotron Plus Information BookletKo Phyo WaiNo ratings yet
- Toxicology ReportsDocument8 pagesToxicology ReportsImam HasanNo ratings yet
- Case Presentation On Chronic Kidney Disease1Document18 pagesCase Presentation On Chronic Kidney Disease1d100% (1)