DEVIATION PROM IOESL-REACTOR PERFORMANCE 297
(0) Write an inital condition forthe acer concentration 2 any locaton within the seater.
{8} Transform the pata diferent sqation abtand in step | (othe Laplace domain. Then sche
the resiking ottinay dierertal mation
(6) fiver the transform solution in scp 40 pve Ci, Ybonhore Cis the concentration of tee ot
time tard ¥ isthe rolume at ans engi the reacor.
(6) Evaluate Ce, V) fo apply tthe ett of the reactor where V = j,. This sves the desired rest,
lh Capes the rr terme af the dene ime @~ 1
1B Steck a pot of C,.Cy 5.1925 prescribed by the slation obtained in A.
. Diseus how avalos fr the eet rato ould be ebeained lor response data: 14 fom
reassert Of wf a sepfction input of ae.
NPrER
SEVEN
HETEROGENEOUS PROCESSES,
CATALYSIS. AND ADSORPTION
HETEROGENEOUS PROCESSES
‘The reaction rates and reactor designs considered thus far have been for hom-
ogencous reactions, The remainder of the book is devoted 10 heterogencous
systems. particularly those involving fluid and solid phases. Such heterogeneous
systems are very important because most chemical reaction processes require a
solid catalyst. Accordingly. the main objective of the next thrse chapters is an
understanding ofthe kinetics of reactions of Muids on solid catalysts. a this first of
the three chapters. the essential differences between homogeneous and heteroge-
vous rates are presented. This is followed by a discussion ofthe general aspects of
catalysis and adsorption. Chapter 8 is concerned with solid catalysts, particularly
theis physical properties, because these properties affect mass and energy transport
catalyst particles. Also included in Chap. 8 is a discussion of theories
of behavior of soled catalysts and practical aspects such as preparation, life, ete
Thea in Chap. 9 we distuss kinetics and rate equations for fluid-phase reactions
‘occurring at the surface of solid catalysts With this preparation we are in pesition
to consider transport effects in heterogeneous processes (Chaps. 10 and 11) and
reactor design (Chap. 13).
There ate leterogeneous reactions other than those involving solid catalysts,
for example, reactions between eases and solids such as in the reduction of metal
lic ores. Such gas-solid, noncatalytic reactions are taken up in Chap. 14. Also,
there are important heterogeneous processes not involving solid phases. Examples
HETEBOCENEOLS PROCESSES, CATALYSIS. AND ADSORPTION 299
are alkylation (liquid-liquid systems) and absorption ard reaction provesses such
1 the separation of CO, and H,S from gases with amine solutions. These kinds of
heterogeneous processes will be considered, but the major emphasis will be on
fluid-solid catalytic reactions
The phase Doundaries inherent in heterogeneous systems requite that trans
port processes (mass and energy transfer) as well as the intrinsic reaction rate be
accounted for in reactor design, Consider the case of a gaseous reaction mixture
‘and a solid catalyst. 1 advantageous 10 use the same conservation equations for
reactor design that were giver. in Chap. 3. Then much of our previous work on
hhomogencous systems (Chaps. 3, 4, and 5) will be apolicable This ean be done if
the reaction rate is expressed in terms of the bulk concentration and temperature
‘of the gas phase. Such cates will include the effects of transport processes. In
Equation (3-1) the rate term is based upon a volume element of reactor. If the
volume element includes a heterogencous region of reaction fiuid and solid eat-
alyst particles. the proper rate wil incorporate massand energy transfer {fom fluid
to solid surface and within the particles (catalyst particles are normally porous).
Such rates are ealled glohal or overall rates and their use allows us to apoly many
of thedesign equations given in Chaps. 3to 5. Ln order to express the global rate in
terms of bulk properties. expressions must be formulated for each of the steps in
the overall process, The sequence of sieps for converting reactants to products is:
|. Transport of reactants from the bulk fluid to the fluid-solid interface (external
surface of catalyst particle)
2. Intraparticle transport of reactants into the catalyst particle (if it is porous)
3. Adsorption of reactants t interior sites ofthe catalyst particle
4 Chemical reaction of adsorbed reactants to adsorbed products (surace
reaction—the intrinsic chemical step)
5. Desorption af adsorbed products
5, Transport of products from the interior sites tothe outer surface of te catalyst
particle
7. Transport of products from the uid-solid interface into the bulk-Auid stream
A: steady state the rates ofall the individual steps will be the same. This equality
‘can be used 10 develop a global rate equation in terms of the concentrations and
temperatures of the bulk fluid. The derivation of such equations will be considered
is detail in Chaps. 10 o 12, bata very simple treatment is given next to illustrate
the nature of the problem,
7-1 Global Rates of Reaction
(Consider an irreversible gas-phase reaction
Ata) Bla)
which requires a solid catalyst C. Suppose that the temperature is constant and
that the reaction is carried aut by passing the gas over a bed of nonporous particles
of C. Since the catalyst is nonporous, steps 2 and 6are not involved. The problem
300 CHEMICAL ENGINEERING KINETICS
is to formulate the rate of reaction por unit volumet of the bad that isthe global
rate fora catalyst particle. r,~in terms of the temperature and concentration of A
inthe bulk-gas stream. Note that those are the quantities tha ere measurable or
‘in be specified (as design requirements) rather than the temperature ard concen-
tration at the gas: particle interface. Global rates of catalytic reactions arc usually
‘expressed per unit mass of catalyst. ie... These are easily converted to rates
Bet unit volume by multiplying them by the bulk density py of the he of catalyst
particiss
The overall conversion of to B in the bulk gas for this case of nonporous
catalyst occurs according to steps 1.3 to Sand 7in series. Let us further simplify
{he problem by supposing that sleps 3 to 5 may be represented by a single
first-order rate equation. Thea the overall reaction process may be described in
three steps: gas transported from the bulk gas to the soli surface. the reaction
cocurs at the interface, and finaly. product B is transported from the catalyst
surface to the Buk gas, Since the action is ireversible, the concenieation of Bat
the catalyst surface does not influence the rats, This mears that, can be for-
mulated by considering only the first two steps involving 4 Since the rates of
these two steps will be the same at steady state the disappearance of A can be
‘expresied in two ways: either as the rate of transport of A tothe catalyst surface,
1s baal —C) (ay
for as the rate of reaction at the eatalyst surface,
Fak, (72)
Jn Eg. (7-1) his the ususl mass-transfer coefficient based on a unit of transfer
surface. ic..a Unit of external arca of the catalyst particle, In order to express the
Fate per unit mass; of catalyst, we multiply k, by the extemal area per unit mass.
nq I Eq, (7-2) & is the reactionrate constant por unit mass of eatalyst, Since a
positive concentration difference between bulk gas and solid surface is necessary
{o transport .t to the catalyst he surface concentration C, will be less than the
bbulk-gts concentration C,. Hence Eq, (7-2) shows that the rate is less than it
would be for C, = C,. Here the elfect of tne mass-transfes resisiance is to reduce
the rate. Figure 7-1 shows schematically how the concentration varies between
bulk gas and catalyst surface.
‘cd such cquarons at
(G-ts)and toujasa are apyeable wo aaMetewtalvolue, kee a poet ate This sn approua
"on whish ais from teatieg the sere nature of abe of eatalyt pacts 2 9 oats
neces approsiation bruise it fot yet posible f take ite acount sarin m heat he
Imassiraufr events with ost onthe sauce oF single catalyst partis. Ths aserage als
‘(m9 oetcrts over the narticesurace areemplored i formulating globulates Th ithe value
dented bY Jia Ea. CH)
$ nstad of ae per unit mas of etl, Eas (1-1) amd (2) cou be eres a asp uit
‘tinal sacs n wish cane ag would mot Be ceded 8 Eg (7-1) agd ki Eg (72) waUld e pe?
lai of catalyst serfice Alemates, arate per particle could be wed. We shall enealy wie he ate
er unit mase
HETEROGENEOUS PROCESS, CATALYS, AND ADSORFTION 301
Cutt tae
Intermediate
eta)
Figure 7-1 Mass transier berwees Mid
ce
‘The global cate can be exprested in terms of C, by first solving for C, from
Eqs. (7-1) and (7-2); thus
(73)
“Then this result is substituted in either Eq. (7-1) or Eq, (7-2) 0 give
Hg 1
Te aR Tk int
“This is the expression forthe global rate in terms ofthe bulkcreaetant concentra-
tion. The second equality shows tha: the effects of reaction and mass transfer are
additive. Since the tems are in the denominator, they are rsitences tothe rae.
“Thus we speak ofa reaction resistance (1/ term) and] a mass-transter resistance
(kad tem), The concentration profile inthis cae is shown by the solid line in
Fig 7-1. 18 a very resticted illustration of a global rate, since heat-transfer
effects were not considered and only external mass transfer is involved (he
lyst particle & nonporous). These restrictions are removed in the detailed treat-
meat in Chaps. 10 and 11, bat this simple example illustrates the meaning of
slobal rates of reaction for heteropencous systems
‘A hypothetical reaction has been used to develop the previous results. It i of
interest to know the magnitude ofthe global rate for real situations, Fortunately
cousderable experimental data ar available. Measutementst fr the oxidation of
$0, with air on a platinum catahst, deposited on } fin. cylindrical pellets,t
gave a global rate of 0.0956 g mol (tg catalyst). The bulk temperature ofthe gas
was 465°C and the gas velocity in the catalyst bed was 360 lbh’). The bulk
‘composition corresponded to an SO; partial pressure of .06 afm. At these condi-
‘bons both mass- and heat transfer resistances between the bulk gas and the sur
face of the catalyst pellets were important. Calculations with Eq. (7-1) indicated
cy. 0-4)
+ R.W.Olion, RW. Schuler, and JM. Sith, Chem Exg. Prop. 2, 614 (1950)
{The parle wore of porn slomina bt the pltinam was deposit onthe outer sacs so
hatin ee, « ronporow east was wed
302 CHESNCAL ENGINEERING KINETICS
thatthe partial pressure of SO, dropped 100040 atm atthe solid surface from the
Value of 0.06 atm in the bulk gas. This relatively lige diference means that the
Gifusion resistance was large. If the rate were evaluated without consideration of
this resistance—-that is, i! it were evaluated for feo, = 0.06 atmat 465°C—It would
bbe 0.333 g moles SO, reacted (hNg catalyst) At pio, = 004 atm it would be
(00730, Henc: the error in neglecting the mass-transfet resistance woul! be large:
thal is, the rate 40 computed would be (0.333.00730)(100), or 350%, higher than,
the eda rate
Actually the temperature at the catalyst surface is about 15°C above the
‘bulk-gas temperature (see Example 10-2) because of heat-transter resistance.
Hence the reaction at the catalyst surface occurred at 480°C, According to the
activation energy for this reaction. a 15°C temperature rise would increase the rate
about 31°, Hence neglecting the heat-iransfr resistance leads toa rate 31%, less
than the global valve. If both thermal and difusion resistances were neglected,
the rate would be (0.333/0.0956)(100) or 250°, higher than the glotal rate. For
this exothermic reaction the diffusion and thermal resistances have opposite
effsts on the rate. The example is extreme in that mass-transer resistance was
relatively huge in comparison with reaction resistance. At lower temperatures
snd or Pier gas velocities past the catalyst pellet (higher &,) the mass-transfer
‘fet would be less
Frequenily external heattransfer resistances are much larger than mass
transfer resistances, in contrast to the preceding example. As an illustration, con-
sider the data of Maymo and Smith? for the oxidation of hydrogen with oxygen
‘ona platinum-on-alumina catalyst, Rates and temperatures were measured for a
‘siagle porous catalyst pellet (1.86 cm diameter) suspended in a well-mixed gas
containing primarily hydrogen with @ small percentage of oxygen and water
vapor. Because of the turbulence inthe gas. the concentration diference between
bulk gas and pellet surface was negligible. However. there was a significant ther-
ral resistance, $0 thatthe pellet surface temperature was greater than the bulk-
sas temperature, The pellet was porous with uniform distribution of platinum
throughout. Hence there were internal resistances. both to mass and to heat
transfer. In one particular run the measured rate of water production was
498 x 10° mol (g catalysi)(s) This is the global value and includes the effects of
both internal and external transport resistances, The bulk-gas, pellet-surface, and
pelle-center temperatures were 899, 101. and 147,7°C. respectively. The rate of
the chemical step on the surface was also measured by experiments with fine
particles of catalyst for which there were no temperature oF concentration differ-
ences between bulk gas and surface oF intctior of particle. Ths intrinsic rate was
correlated by the equation
= 0655 182%
where pis in atmospheres and Tis in degrees Kelvin. We can evaluate the effect of
the external resistance inthe following way. First calculate the rate atthe surface
temperature (LOL +273 K) and po, = 00527 atm, which is the oxygen pactial
susen7 (v5)
1A. Mayme and J. M-Simiih, AFORE J. 52 85 (1966)
ETEROGENEOL'S PROCESSES. CATALYSS, AND ADSORPTION 303
pressure, ether in the bulk gas orat the pellet surface. Substituting these quanti
ties in Eq, (7-5) gives e, = 54.3 x 10°? mol (g catalyst)(6). If the rate were eval-
ated at bulk-gas conditions (899°C and fo, = 0.0527 atm), the result from
Eq, (7-5) would be 436 x 10°® mo! (g catalysts). Hence neglecting the external
temperature difference would give a rate ((S43 — 43.6543] 100), of 20% less
than the correct value.
“This example also shows the effects of mass- and energy-transie: resistances
‘within the catalyst pellet. The temperature increases toward the center of the pellet
and increases the rate, bt the oxygen concentration goes down, tending to reduce
the rate, The global value of 493 x 10°* is the resultant balance of both factors.
Hence the net error in using the bulk conditions to evaluate the rate would be
[149.8 ~ 43.6),49.8](100), 12.5%, In this ease the rate increase duc to external and
internal thermal efects more than balances the adverse efiect of internal mass-
transfer resistance, The procedure for calculating the eflcts of internal gradients
fon the rate is presented in Chap. 11
One additional illustration is of interest. This refers to the hydrofluorination
of UO, pellets with HF(g} sccording to the reaction
UO, (0) + 4HF |g) + UF 6) + 21,019)
“This ie a diferent kind of heterogeneous reaction—a ges solid noncatalytic ons.
Let us examine the process at initial conditions (¢ 0), s0 thet there has been no
‘opportunity for a layer of LIF,(s) to be formed around the UO, pellet. The
process is much like that for gas sold catalytic reactions. Hydrogen fluoride as is
transferted from the bulk gas to the surface of the UO. pellets and reacts atthe
pellet-288 interlace, and H,0 diffuses out into the bulk gas Ifthe pellet is nox-
Porous, al the reaction oesurs at the outer surface ofthe UO; pellet. and only an
external transport process is possible. Costa® studied this system by suspending
spherical pellets 2m in diameter ia a stired-tank reactor. In one run, at a
bulk-gas temperature of 377°C, the surface temperature was 462°C and the ob-
served rare was r= 69 x 10-* mol UO; (s)(om? reaction surface}. At these
conditions the concentrations of HF gas were 1.12 x 10-* g mol/em? at the sur-
face ofthe pellet and 1.38 = 10° g mol‘? in the bulk gas, The intrinsic rate of
reaction was found to be given by
F = 40Cyp2°6°7 47 g mol/(ern?}(s) (7-6)
where T isin degrees Kelvin and Cy is in gram moles per cubic centimeter. Ifthe
rate is evaluated at bulk-gas conditions, Fq, (7-6) gives 50 x 10-° g mol(s)iem"),
‘Then the combined effects of an external temperature and concentration differ
tence serve lo increase the rate from 50 x 10°* to 69 x 10-° g mol/(em*)s). or
38%. In this case the temperature at the catalyst surface fs 95°C higher than the
bulk-gas temperature, and this has a dominating effect on the rate of reaction. The
fleet of external mass-iransler resisiance is tO reduce the rate of this first-order
reaction by (1.38-— 1.12)/1.38, or 19°,
EC Conta and J, M. Smith, AICAEY. 17,987 (3971)
304 CHEMICAL ENGINEERING KINETICS
7-2 Types of Heterogeneous Reactions
All ehe examples in See. 7-1 were of the gas-solid form, As mentioned carlir this is
‘a most important type of heterogeneous system. Most salable chemicals are
prepared by converting raw materials via chemical reactions, Such reections
usually require catalysis, and these are normally solids. Since the temperatures
must be high for rapid cates, the reacting fluid commonly is in the gas phase.
Examples of large-scale gas-colid catalytic reactions are the major hydrocarbon
transformations: cracking. reforming, dehydrogenation (for example, butadiene
and butenes from butane}. isomerization, desulfurization, ete. Kinetics and reactor
design for such systems have naturally received major attention, and most of our
emphasis on design is directed to this type of Row reactor (Chap. 13),
Gas-solid heterogeneous reactions may be noncatalytic.* An example is the
hydrofuorination of uranium dioxide pellets referred to in Sec. 7-1. Since one
reactant is in the solid phase and 's consumed, the rate of reaction varies with
time. Hence such processes are intrinsically dynamic, in comparison with the
stead-state operation of gas-solid catalytic reactors. The process for smelting oret
‘such as zine sulfide,
ZnSls) + 30,(9) + Zn0s) + SO.)
'S of this type. The conversion of CaCO to CaO in the live kiln is another
example, Yet another is the process for making HCI in a transport reactor’ irom
salt particle: the reaction is
2NaCi(s) + SOG) + HyO(g)-+ Na,SO4(s) + 2HCI(a)
In many noncatalytic types a sold product builds up around the reacting core [for
«exarnple, Na;S04(5) is deposited around the NaCI particles in the last ilustra-
tion). This introduces the additional physical processes of heat and mass taster
through a prodluet layer around the solid reactant. somewhat diferent forn of
noncatalytic 235 selid reaction is the rogeneration of catalysts which have Seen
deactivated by the deposition of a substance on the interior surface, The most
common isthe burning (with air) of carbon which has been gradually depesited
on catalyst particles used in hydrocarbon reactions. Many of the physical and
chemical steps involved here ave the same as those for gas-solid catalytic reactions.
The chit dillerence the transient nature of the roneatalytic reaction. Ths type
of heterogencous reaction will be considered in Chap. 14, Another dynamic a0n-
catalytic process ithe separation or purification of gases or liquids by adsorption.
Such processes are normally carried out in tubes packed with the adsorbent
partices, Adsorption of the higher molecular weight hydrocacbons from gas
streams in beds of activated carbon and removal organic pollutants fom water
with setivated carbon are examples.
‘Jian Saskely. James W. Eeane aed Hong Yong Soha," Ga-Solid Reactions” Academie Pit
Sew York 1976
A teaspor:ceactor ikea Muidzedsbed reactor (Fig, 8). except that the Muidzed partes
move through and out ofthe reir with the ass phase
HETEROGENEOUS PROCESSES, CATALYSES, AND ADSORPTION. 30S
A steady-state example of noncatalyic reactions is the gas-liquid form in
which a gascous reactant is absorbed in the liquid and subsequently reats i the
liquid phase. The eeactor in suck cases is usually © countercurrent or cocurte
absorption column of the bubble type’ (gas bubbling through a continuous liquid
phase, spray column or a tube packed with inert particles). Examples are the
separation of CO; from a gas by adsorption and reaction in an aqueous amine
solution t
Liguid-solid reactions, where the soli phase i a catalyst, isa type of heter~
‘ogencous system that is common i the chemical and petroleum industries. Alkyl-
ation with AICI,(0) catalyst is an example. In these systems the catalyst frequentiy
forms complexes with reactants and/or products and becomes a poorly defined
solid-liquid mixture, best described as a sludge. Analytical treatment in these cases
is dificult, Three-phase systems involving a gs, liguid, and solid are also en-
countered. Hydrogenation of liquids normally is of ths type. For example. a
slurry may be formed of the catalyst particles inthe el to be hyerogerated. Then
tydcogen is bubbled into the slurry. These reactions are carried out in sttred-tank
reactors, where the high heat capacity ofthe tank contents simplifis temperature
control of the exothermie hydrogenation reaction, Such gas-liguid-slid systems
involve several physical steps, and indeed, resistance 10 diffusion of dissolved
hydrogen to the catalyst paticle is frequently significant. The quantitative treat-
ment of combined physical and chemical steps in such slurry reactors is con-
sidered in Chape. 10 and 13, Polymerization systems are often of this type. For
example, ethylene is polymerized by dissolving it in a solvent containing sus-
pended catalyst particles. The trickle bed reactor is a somewhat different form of
gas-liquid-solid system. Here gas and liquid. usually ia cocurrent flow, pas over a
bed of catalyst particles in a fixed bed. This form is used when the volatility ofthe
liquid is so low that complete gasification is nct practical. Sultur compounds are
removed from hydrocarbon liquids in such reactors. The gas phase is primarily
hydrogen. Oxidation of liquids aso is carried out in trckle-bed reactors (see
Chaps. 10 and 13)
Liquid-liquid reactions are sometimes encountered. Alkylation of hydrocar-
bons wih sulfuric acid or HF as a catalyst is an example
‘Solid solid noncatalytc reactions are important in ceramics manufacture § It
appears that diffusion resistances may be important to some extent in all such
systems: The diffusion process ieee diffeult to define in toid-toid systems. At
{east two possibilities erist: volume difision in the sold and surface difusion
aloog interfaces and crystal boundaries € Litle is known about the kinetics of
Solid-soli reactions atthe reacting interface because most measurements include
difusion effect.
F.G. Hagherg and F.X. Krupa, Proc. 4th Im. Simp, Chem. Rest Em. See 1X. p48, Api
1916. Heideerp DECHEMA, Franifurt
YP. V. Danckwers" Gas Ligud Reactions” McGraw Hil Back Company, New Yer, 194.
3. Kingery, "Kinetis of High Tenperature Precsss.” Join Wiey & Som. inc, New York,
1959: 6. Cohn. Chem. Re. 42. $27 (1948)
"RD, Arvoameith and’) M. Sith. Pd, Eng Chom. Fund, Quart. 327 (1968)
306 CHEMICAL FNGINEERING KINETICS
CATALYSIS
“The fest step in determining global rates is to etamine the processes associated
swith the solid surlace (the aésorption, desorption, and intrinsic reaction steps in
the Ist at the beginning of the chapter) We start out with a brief qualitaive
iscwsion of the nature of catalysis and then complete this chapter with an
examination of adsorption. As kinetis information began to accumulate during
the lust century, it appeared that the rates of a number of reactions were
influenced by the presence of a material which itself was unchanged during the
process. In 1836 Berzeliust reviewed the evdence and conciuded that a “cata
Iytie™ force was in operation. Among the cases he studied were the conversion of
siarch into sugar in the presence of acids, the decomposition of hydrogen peroxide
in alkaline solutions, and the combination of hydrogen and oxygen an the surface
of spongy platinum, In these three examples the acid. the alkaline ions, and the
spongy platinum were the materials which increased the rate and yet were vit-
ually unchanged by the reection. Although the concept of a catalytic force
proposed by Rerzehux has now heen discarded. the term “catalysis” is retained 10,
Gescribe all processes in which the rate ofa reaction i influenced by a substance
that remains clemically unaffected.
7-3 The Nature of Cat
Iytic Reactions
Although the catalyst remains unchanged at the end of the process, there is no
requirement that the material not take part in the reaction. In fact, present
theories of catalyst activity postulate that the material does ectively participate in
the reaction. From the concept of the energy of activation developed in Chap. 2.
‘the mechanism of catalysis would have to be such that the free energy of activation
is lowered by the presence of the catalytic material. A catalyst is effective in
increasing the rate of a reaction because it makes possible an alternative mechan-
ism, each step of which has a lower free energy of activation than that for the
luncatalyzed process. Consider the reaction between hydrogen and oxygen in the
presence of platinum, According 10 the proposed concept. hydrogen combines
‘with the platinum to-form an intermediate substance, which then reacts with
oxygen to provide the final product and reproduce the catalyst. tis postulated
that the steps involving the platinum surface occur at a faster rate than
the homogeneous reactions between hydrogen and oxygen.
‘The combination or completing of reactant and catalyst a widely accepted
basis for explaining catalysis. For example, suppose the overall reaction
ABC
is catalyzed via two active centers, or catalytic sites, X, and Xz, which form
‘complexes with 4 and B. The reaction is traly catalytic ifthe sequence of steps is
113 Borveoe leche, Chem. 1.237 (1836)
ETEROCESEOUS PROCESSES CATALYSES, AND ADSORETION 307
such that the centers X', and X, are regenerated ater they have caused the
formation of C, In a general way the process may be written
1 ASX, AN,
2 B+ X,=BN,
BAN EBXVEC HN HMD
Note that whereas X, and X are occupied and regenerated a number of times,
does not necessarily follow that their catalyzing ability and/or number remain
constant forever. For example. poisons can intervere tostowly remove X and/or
X; from the system. arresting the catalytic rate. What distinguishes this decline in
catalytic activity from that of a noncatalytic reaction in which X; and Xpare not
regenerated is that the complexing-regencrating sequence occurs a great many
times before Xy and 2 become inactive. In the noncatalytie sequence no regener~
ation of X occurs. Hence. while catalysts can deteriorate, thei active lifetime is fur
jteater than the time required for reaction,
‘A relatively small amount ofeatalyst can cause conversion of a larg: amount
of reactant. For example, Glasstone* points out that cupric iors in the concentra-
tion of 10°? mol liter appreciably increase the rate of the oxidation of sodium
sulfide by oxygen, However. the dea that a small amount ofthe catalyst can cause
a lerge amount of reaction dors rot mean that the catalyst concentration is
unimportant. In fact. when the reaction does not entail a chain mechanism. the
rate of the reaction is usually proportional (0 the concentration of the catalyst
‘This & pethaps most readily understood by considering the case of surface eata-
Iytic reactions. Inthe reaction of hydrogen and oxygen with platinum catalyst the
rate is frequently directly proportional to the platinum surface. Here a simple
DProportionality exists between platinum surface area and the number of centers X
which catalyze the oxidation of hydregen. While such a simple relationship may
not often exist in solif-catalyzed reactions. in homogencous catalysis there 1s
frequently a direct proportionality between rate and catalyst concentration. For
‘example. the hydrolysis of esters in an atid solution will depend on the concentra-
tion of hydrogen ion acting as a catalyst.
‘The position of equilibrium in a reversible reaction is not changed by the
presence of the catalyst. This conclusion fas been verified experimentally in
several instances: For example, the oxidation of sulfur dioxide by oxygen has been
studied with three catalysts: platinum, feric oxide, and vanadium pentoxide. In
all three eases the equilibrium compositions were the same.
‘An important characteristic of a catalyst iit effect on selectivity. An illustra
tion is the decompesition of ethanol. Thermal decomposition gives water, acetal:
dehyde. ethylene. and hydrogen. If, however, ethanol vapor is suitably contacted
with alumina particies.ethylene and waterare the only products. In contrast dehy:
drogenation to acetaklchyde is virtually the sole reaction when ethanol is reacted
‘over a copper catalyst
#5. Glastone "Teubook of Physieal Chomistey"p 1104, D. Van Nowrand Company tee, New
You. 190,
308 CHEMICAL ENGINEERING KINETICS
‘The general characteristics of catalysis may be summarized as follows:
| A catalyst accelerates reaction by providing alternate paths to products, the
activation energy of each catalytic step being less than that for the homoge-
neous (noncatalytic) reaction.
2. In the reaction cycle, active centers of catalysis first combine with at least one
reactant and then are reproduced with the appearance of product. The freed
‘center then recombines with reactant to produce another cycle, and so on.
3. Comparatively small quantities of catalytic centers are required to produce
large amounts of product.
4. Equilibrium conversion is not altered by cetalysis. A catalyst which accelerates
the forward reaction in an equilibrium system is a catalyst for the reverse
reaction,
5. The catalyst can radically alter selectivity
Examples have teen observed of negative catalysis, where the rate ie de-
‘creased by the catalyst. Perhaps the most reasonable theory is that developed for
chain reactions. In these eases it postulated that the catalyst breaks the ceaction
chains, or sequence of steps, involved inthe mechanism. For example. nitric oxide
restuces the rate of decomposition of acetaldehyde or ethyl ether. Apparently
nitnc oxide Fas the characterise of combining with the ree radials involved in
tke reaction mechanism. The halogens, particularly iodine, also act as negative
catalysts in certain gaseous reactions. In the combination of hydrogen and
oxygen, where a chain mechanism is probebly involved, iodine presumably de-
Stroys the cadicals necessary forthe propagation cf the chains
Reactions in which the raz increases with product concentration also occur
Such easton are termed autocatalyte. The most sommon examples ae fermen
tation processes “catalyzed " by microoreanisrs such as yeasts, bacteria. and
algae. The peocesies ars compl but maybe described approximately a a ceac-
tion between an organi fed and microorganism to produce produets and more
‘microorganisms. For example, the fermentation of augar may be expressed:
Organic feed + microorganism -+ inert produets + more yeast (7-7)
(ee po) eas) (akoneiy
Hf batch reactor initially contains a sugar solution and a small quantity of yeast,
‘he rate will increase as the reaction proceeds, because the Yeast produced is also a
reactant, Ultimately. the depletion of sugar will cause the rate to reach a maxi-
‘mum and decrease toward zero. The microorganism does not act asa true catalyst
since it is a reactant, but its presence does increase the rate, Such abnormal
‘kinetics has been extensively treated in the literature. partly because the unusual
rate v5. conversion relationship leads to interesting problems of reactor choice and
‘optiraization.*t The kinetic equations are illustrated in Prob. 7-5 for bacterial
oxidation
+.B Bacholl Con J. Chom Erg. 4,26 (1966)
$1 F Lele, “Chemis! Kintion” MeGraw-Hil Book Company, New York, 1965.
HETEROGENEOUS PROCESSES, CATALYSIS, AND AESORITION 349)
7-4 The Mechanism of Catalytic Reactions
‘The concept that a catalyst provides an alternate mechanism for accomplishing a
‘reaction, and that this alternate path is a more rapid one, has been developed in
many individual cates, The basis for the theory is that fist the catalyst and one or
‘more of the reactants form an intermediate complex, a loosely bound compound
which is unstable. This complex then taker part in subsequent reactions which
result in the final products and the regenerated catalyst. Homogeneous catalysis.
can Irequently be explained in terms of this concept. For example, consider catal-
ysis by acids and bases. In aqueous solutions acids and bases can increase the
rate of hydrolysis of sugars, starches, and esters, The kinetics of the hydrolysis of
ethyl acetate catalyzed by hydrochiorie acid'can be explained by the following
mechanism:
1, CH,COOC,H, + H” =CH,COOC,H,[H*]
2, CH,COOC,H,[H"] + H,0=:C,H,OH + H* + CH,COOH
For this catalytic sequence to be rapid with respect to noncatalytic hydrolysis, the
free energy of activation of reaction steps 1 and 2 must each be less than the free
‘energy of activation for the noncatalytic reaction,
CHyCOOC;H, + 1,0 =CHCOOH + CH,0H
Similarly, the heterogencous catalytic hydrogenation of ethylene on a solid
‘catalyst might be represented by the steps.
6
1 GHex, Zee,
2 HGH, SCH IH
3. CHALK JH, HOH +X)
where X, 8 an active site on the sod catalyst and CH[X,]H, represents the
complex formes between the reatanis and the catalyst, The homogeneous Teag-
tion, according to the absolute theory of reaction tates dsctssed in Chap. 2
would be writen
cit
an Comper
CHL + Hy SOM Gal,
where the free-energy change for the formation of the activated complex AFY is
the free energy of activation for the homogeneous reaction. The effectiveness of the
catalyst is explained on the basis that the fre energy of activation of each of the »
steps in the catalytic mechanism is less than AF*,
These illustrations, particulary that for ethylene hydrogenation, ace grossly
oversimplifiod. They must be considered phenomenological models, not mecha-
fisms. The actual mechanism of ethylene hydrogenation is quite complex. In spite
of the considerable effort focused on this reaction, a mechanism satisfactory to all
310 CHEMICAL, ENGINEERING KINETICS
investigators has yet to be offered. The system dots, however, provide an oppor-
tunity to compare homogeneous and heterogeneous rates. Using published data.
Boudert* found that the homogencous and cataltic rates can be expressed as
hon = 10 A806 08)
ey = 2 x 1087019800 HT (CUOMO catalyst) 0-9)
AL 600 K the relative rates are
fot = gtst0o-tsapnton 49!
Inthis case the catalyst has cause 2 radical reduction in overall activation energy.
presumably by replacing a difficult homogencous step by a more easily execuced
surface reaction involving adsorbed ethylene. The results lead to the kinetics
observed by Wynkoop and Wilhelm.t a reaction frst order in Hy and zero order
in strongly adsorbed ethylene
‘The three steps postulated for the catalytic hyérogenation of ethylene indicate
that the rate may be influenced by both adsorption and desorption (steps 1 and 3)
and the surface reaction (step 2), Exireine cases can de imagined: that step | or
step 3is slow with respect to step 2 or that step? is relatively slow. In the fist wo
«eases cates ofadsorption or desorption ate of interest since they determine the rate
while in the chird case the surface concentration of the adsorbed species, corre-
sponding to equilibrium with respect to steps 1 and 3, & needed. In any event we
should like to know the number of sites on the catalyst surface. or at leas the
surface area oft vataiyst. These questions require a study of adsorption equilib-
rium and adsocption rates. This isthe objective in Secs. 7-5 t0 7
The detailed mechanism of how solid catalysts operate requires knowkedge of
the structure ofthe complex between reactant and catalyst site Understanding of|
this aspect to catalysis has been slow in developing. A brief review of suggested
theories and experimental evidence is given in Ses. 8.4
ADSORPTION
7-5 Surface Chemistry and Adsorption’
Even the most carcfuly polished surfaces are not smooth in a microscopic sense,
but are irregular with valleys and peaks alternating over the area. The regions of
ieregularity are particularly susceptible to residual force flelds. At these locations
the surface atoms of the solid may attract other atoms or melecules in the su-
rounding gas or liquid phase. Similady, the surfaces of pure crystals have noauni-
‘$M. Bondar, fl Chm Dlg, 29.383 (1958,
ER Wynkoop ard R. H. Wilhelm, Chem. Ey, Progr a, 0 (1959)
SA dicision ofthe thermedjnamics ed kinetics of abortion pen by ed Clark “The
‘Theory af Adsoeption und Catalysis” Academie Prom, Sow York, 1979
HETEROGENEOUS PROCESSES. CATALYSIS, AND ADSORPTION 311
form force fields because of the atomic structure in the erystal. Such surfaces also
have sites or active cemie's where adsorption is enhanced. Two types of adsorption
may occur.
Physical adsorption* Physical adsorption is nonspecific and somewhat similar to
the process of condensation. The [orees attracting the fluid molecules to the solid
surface are relatively weak, and the heat evolved during the exothermic adsorp.
tion process is ofthe same order of magnitude as the heat of condensation, 0.5 to 5
kcal/g mol. Equilibrium between the slid surface and the gas molecules is usually
rapidly attained and easly reversible. because the energy requirements are smal.
“The energy of activation for physical adsorption is usually no more than 1
salig mol, since the forces involved in physical adsorption are weak. Physical
adsorption cannot explain the catalytic activity of solids for reactions betwen
relatively stable molecules, because there is no possibility of large reductions in
setivation energy. Reactions of aloms and free radicals at surfaces sometimes
involve small activation energies, and in these eases physical adsorption may play
0 sves t0 concentrate the molecules of a sub-
stange at a surface.‘This ean be of importance in eases involving reaction beta
a chemisorbed reactant and a coreactan,which can be physically adsorbed in such
4 system the catalytic reaction would otcur between chemisorbed end physically
adsorbed reactants
Te amount of physical adsorption decreases rapidly 2s the temperature i
raised and generally very small above the critical temperatures of the adsorbed
component. This is further evidence that physical adsorption isnot responsible for
saialysis. For exampk. the ralc of oxidation of sulfur dioxide on a platinum
ttalyst becomes appreciable only above 300°C. yet this considerably above the
critical temperature of sulfur dioxide (157°C) or of oxygen (—119°C), Physical
adsorption is not highly dependent on the irregularities in the nature of the
surface, but i usually diect!y proportional to the amount of surface. However.
the extent of adsorption is not limited to a monomolecular layer on the solid
Surhice, especially near the condensétion temperature. AS the layers of moleciles
Build up on the solid surface, the process becomes progressively more like one of
condensation,
Physical-adsorption studies are valuable in determining the physical proper:
ties of solid catalysts. Thus the questions of surface area and pore-ize distribution
in porous catalysts can be answered irom paysical-adsorption measurements
‘These aspects of physical adsorption are considered in Chap 8.
Chemisorptiont The second type of adsorption is specific and involves forces
much stronger than im physica! adsorptiony According to Langmuir’s pionecr
work the adsorbed molecules are held to the surface by valence forces of the
+ Fora dates (reaimen’ of physical adsorption see D.M. Young and A. Crows “Physial
Adsoepion of Gases” Botierworth & Co. (Publishers). London, 1962
{Fora deta (veatmon of chemisorption ste DO. Hayeard and BM, W. Tropa” Chem
soxpiicn.* 2d e@. Butterworty & Co, (Publishers, London, 1958
ԤLLangmuie Jam Chem S00, 38 2111936)
32 CHEMICAL ENGINEERING KINETICS
sume type as those occurring betwsen atoms in malecules, He observed that «
stable oxide film was formed on the surface of tungsten wites in the presence of
‘oxygen, This material was not the normal oxide WO, . because it exhibited differ.
‘ent chemical properties. However, analysis of the walls of the vessel holding the
wite indicated that WO, was given off from the surface upon desorption. This
suggested a process of the type
30, + 2W + 2[W-0,]
2[W-05] +2W0,
where [W-Os] represents the adsorbed compound. Further evidence for the
theory that such adsorption involves valence bonds is found in the large heats of
adsorption. Observed values are of the same magnitude as the heat of chemical
reactions, 5 to 100 keal’g mol.
Taylort suggested the name clmisorption for describing this second type of
combination of gas molecules with solid surfaces. Because of the high heat of
adsorption. the energy possessed by chemisorbed molecules can be substantially
atocpte
1 has been explained that the effectiveness of solid catalysts for reactions >
stable molecules is dependent upon chemisorption. Granting this. the temperatare
ange over which a given catalysts effective must comeide with the range where
chemisorption of one of more ofthe reactants is appreciable. This is indicated 02,
Fig. 7-2 by the dashed vertical lines. There is 2 relationship between the extent of
chemisorption of a gus on a solid and the effectiveness of the solid as a eatalys
For example, many metalic and metal-oxide suriaces adsorb oxygen easily, and
these materials are also found to be good catalyst, for oxxlation reactions. Whe
feactions proceed catalytically at low temperatures, Fig. 7-2 does not apply.
these eases catalysis & due to nonaeticated chemisorption. Thus ethylene i hy
rogenated on nickel at — 78°C, at which temperature there would surely eu
physical adsoeption of the ethylene.
An imporiant feature of chemsorption is that ils magnitude will not exee0
‘that corresponding to a monomolecular layer. This limitation is due to the fact
‘that the valence forces helding the molecules on the suzface diminish rapidly wits
itance. These forces become too sinall to form the adsorption compound wher
the distance from the surface is much greater than usual bond distances
The diflcieacss between chemisorption and physical adsorption are sur
marized in Table 7-1
In order to des elop eats equations for catalytic reactions quantitative expres
sions for adsorption ate necessary. Langmuir? proposed simple formulations for
rates of adsorption and desorption of gases (applieable also t0 liquids) on solid
‘surfaces, While his concern was with chemisorption. the concepts have been uscd
for deriving a valuable relationship between the volume of a gas physically ad.
sorbed and the surface area of the sold (see Sec, 8-1). Applied to chemsorption.
Langone Js don Che Si, 11 (1915.
314 CHBNICAL ENGINEERING KINETICS
Table 7-1 Physical vs. chemical adsorption
Peamaee Prysalagsorqtion —_ Chemompuon
‘horton A elise Some sods
soctte All ees low ‘Sonne chemeally
‘teal empe reasive pes
Temperature eange Low temperatere Generally igh
Ha of abortion Low (2AM High order ot test,
ate. acination erergy Vey raph low E—Nonaaivatd, lew E:
stated gh €
‘cae Mattia posible Movolayer
Reversi Nights rverible Oem erenere
Iroportance For determination of For determination of
ace neta and Tearicpenle arena
pore sue ‘toazton ots
fesstion kin
‘the Langmuir ircatment provides a systematic basis for developing rates of eier-
‘geneous catalytic reactions and we vse this approach in Chap. 9. Adsorption and
ecorption steps in the everall conversion of reactants to products (see list at
ning of this chapter) may be very fast and hence, occur at near equilibrium,
“Therefore. we need also to know the equilibrium relation between the concentra
tion of adsorbent ia the gas (of liquid) and the amount adsorbed, Such equili-
brium isotherms. Langmuir and other forms, are considered in Sec. 7-6 and then
rates of adsorption are discussed in See, 7.7
7-6 Adsorption Isotherms
A. The Langmuir treatment of adsorption ‘The derivations may bé carried out by
Using as a measure of the amount adsorbed either the Faction of the suriace
cowered of the concentration ofthe gas adsorbed onthe surface Bath procedures
willbe illustrated. although the second is the mere useful for kinelx developmenss
(Chap.9) The important assumptions areas follons:t
J. All the sustuce of the catalyst has the same aetvity for adsorption; ve. it is
cnetgetically uniform. The concept of nonuniform surface with active centers
can be employed iF it assumed that all the active centers nave the same
activity for adsorption and that the rest of the surface has none, or that an
average activity can be used
There is no interaction between adsorbed molecules. This means that the
amount adsorbed has no effect on the rate of dsoeprion per site
1 & sho suppose tht sich ke ca ascommadate any one adeoebt pat
ETIROCENEOUS PROCESSES, CATALYSIS, AND ADSORPTION 31S
3. All the adsorption occurs by the same mechanism, and cach adsorbed complex
has the same structure
4. The extent of adsorption is less than one complete monomolecular layer on the
surface.
In the system of solid surfate and gas, the molecules of gas will be continually
striking the surface and a fraction of these will adhere. However, because of cheit
kinetic, rotational, and vibrational energy, the more energetic molecules will be
continually leaving the Surface. An equilibrium will be established such that the
rate at which molecules strike the surface, and remain for an appreciable length of
time, will be exactly balanced by the rate at which molecules leave the surface.
“The rate of adsorption r, will be equal to the rate of collision r, of molecules
with the surface multiplied by a factor F representing the fraction of the colliding
molecules that adhere. At a fixed temperature the number of collisions will be
‘proportional to the preseure p of the gas (or its concentration), and the fraction F
‘will be constant. Hence the rate of adsorption per unit of bare surface will ber, F.
‘This is equal to kp, where k is a constant involving the fraction F and the propor.
tionality between r, and p.
‘Since the adsorption is limited (0 complete coverage by a moresnolecular
layer, the surface may be divided into two parts: the fraction 0 covered by the
adsorbed molecules and the fraction I~ 6, which is bare. Since only those
molecules stricing the uncovered part of the surface can be adsorbed. the rate of
adsorption per unit of total surface will be proportional to 1 ~ 0: that i,
t= kp(l — 0) (7-10)
‘Tho rate of desorption will be proportional to the fraction of covered surface
ako oa)
“The amount adsorbed at equilibriuin is obtained by equating r, and r, and solving
for 6. The result, called the Langmuir isorhierm. is
be. Kp.
Ge om AP
=e TH Kp
where K =k is the adsorption equilibrium constant, expressed in units of
(pressure)™', The fraction 0 is proportional to volume of gas adsorbed. since the
adsorption is less than a monomolecular layer. Hence Eq, (7-12) may be regarded
a5 a relationship between the pressure of the gas and the volume adsorbed. This is
indicated by writing d = tt, where ¢, ithe Volume adsorbed when all the aciive
sites are covered, ic.. when there is a complete monomolecular layer.
The concentration form of Eq, (7-12) can be obtained by introducing the
concept of an adsorbed concentration C. expressed in moles per gram of catalyst
IFC, represents the concentration cortesponding to a complete monomolecular
tae’ on the cat. then the rate of adsorption, cya ctl) by snags
with Eq, (7-10).
tee bellu 0) (3)
316 cueMieaL ENGINEERING KINETICS
where kis the rate constant for the eatast and C, is the concentration of
sdcorbable component in the gas. Similarly. Eq. (7-11) becomes
ake 14)
AL equilibrium the rates given by Eqs. (7-13) and (7-14) are equal, so that
KiGacy
CST eG
vis)
where now the equilibrium constant K, is equal to k, Ke and is expressed in units
fof concn}“*,centimeters per gram mole. Since CC, = 9.Eq. (7-15) may also be
which ie a form analogous to Eq. (7-12). since C, is proportional to p
‘Equation (7-15) predicts that adsorption data should have the general form
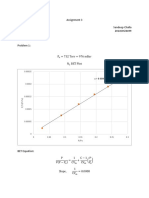
shown in Fig 7-3, Note that at low values of C, (or low surface coverages 6) the
{expression becomes a straight line with a slope equal to K, Cy. The data points in
Fig. 7-3 are for the physical adsorption of m butane on silica get (S, = $32 m/e)
at $0°C+ The solid line represents Eq, (7-15). where
Gy = 085 «10°? g mig silica gel
and the equilibrium constant of adsorption is
K,= 4.1 « 10° emg mol
For this instance of plpsical adsorption Eq. (7-15) fits the data rather well The
measurements wece made on mixtures of m-butane in helium (at 1 atm total
pressure) t0 vary C,. The percentage of n-butane in the gas corresponding to the
conesntration C, is shown as a second abscissa in the figure. Alko, Oi shown asa
second ordinate’ This was calculated from 0 = CC, Its interesting tonote that
the isotherm is linear up to about = 0.10, ora gas concentration of 1", n-butane
in He, This means that the denominator in Eq. (7-15) is nearly unity at low
concentrations. The region of linearity depends upon the adsorbent-adsorbate
system. For example, for the same silica gel at 30°C, the linear isotherm was
applicable up to 2”, for adsorption of propane and to more than 4*, for ethane:
‘Chemserption data often do not ft Eq, (7-15). Howerer, the basic concepts
fon which the Langmisic isotherm is based (the ideas of a dynamic equilibrium
‘between rates of adsorption and desorption and a finite adsorption time) are
sound and of great value in developing the kinetics of fiuid-solid catalytic reac-
tons. Equations (7-13)to (7-15) form the basis for the rate equations presented in
Chap. 9.
+P. Sohnsiter snd JM Sith, ACME J. 14 789 (1968)
NETEROGENEELE PROCESSES, CATALYSIS. AND ADSOEPTION 317
wo eas
0s T T 7
. © Fave
S| SHS das
= os
+— :
i i
: | | 2
hoo a B
z Lier stem | 3
: 4
é | 5
2 z
po f £
[es
aes ie 30
Gas concentration, C, * 10%. molicm?
Figure 7-3 Adsornton-equiftium data for w-batane on slept surface area 832 mg) at SOC
B, Other isotherms and heats of adsorption Experimentally. the five types of iso
therms shown in Fig. 7-4 have been observed +t Note that the linear form of
Eq. (7-19)
Cm AK CaIC, (77)
‘Would fit all ofthe five isotherms at very low fuid concentrations. However, only
eurve | agrees with the Langmuir expression over the whole concentration range
‘The deviations from the Langmuie form are probably largely due to the postulates
‘of sites of equal activity and no interaction between occupied and tare sites. I
right be surmised thet these assumptions correspond to a constant beat of
Adsonpiior. Indeed itis possible to derive the Langmuir isotherm by assuming
that AH, is independent of 0. The heat of adsorption can be evaluated fot
‘dsorption-equilitrivm data, First the Clausius-Clapeyton equation is applied to
the two-phase syslem of gas and adsorbed component-on the surface
(#)-artm
+ D.O Maynard and BM. Trapnell, “Chomsiorption” 34 ed Buttrnorth & Co. (Pate
5). London. 1958
Mtns J. Piss Chom, $3,712 (1999)
1B CHESHCAL ENGEMEERING NINETIES
Amount adsorbed o>
Amount absorbed, C—
Figure 74 Types of adsorption others
where Vand ¥, are the volumes per mole of adsorbed component in the gas and
fon the surlace. respectively. Neglecting the latter and assuming the ideal-gas law
for V gives
was)
For the Clausius-Clapeyron equation to be valid the process must be univariant,
‘This means that Eq. (7-18) can be applied only for constant concentration of
adsorbate on the suriace, that is, constant 0, I adsorption-cquilibrium data are
available at diferent temperatures, the slopes of pvs.-T curves at constant @ may
be used with Eq, (7-18) to caleulate AH, . Figure 7-5, taken from Beeck,* shows,
‘such isosteric heats of adsorption as a function of for hydrogen on several metal
films. These results are typical in showing a decrease in AM, with increasing,
surface coverage, For physical adsorption, AH, values are lower For example. for
nitrogen and »-butane the heats of adsorption (at essentially zero surface cover
‘age) on a molecular sieve catalyst (synthetic zeolite, SA type) were 43 and 103
kealig mol.
‘Two other wellknown wotherms may be clasifed in terms of the AF, ~ 0
dependency. The Temkin isrfierm may be derived from the Langmuit isotherm
10. eck, Dae Faraday Soe 6. 118 1950)
EN Hashimoto and 3M Smith al By. Chor, Funda, 12 383973)
HETEROGENEOUS PROCESSES, CATALYSIS, AND ADSORPTION 319
by assuming that the heat of adsorption drops linearly with increasing 0.1 The
result is
Oak Inkp (7-19)
where ky and k, are constants at a given temperature. Figurp 7-5 shows that a
linear decrease in AH, with 9 fits some data for H on metal films. Similar
agreement has been obtained for N; chemisorbed on a promoted iron catalyst.
“The Freuntlich sovierm can be derived§ by assuming a logarithms decrease
in AH, with 0; that i,
AH, = ~AHe In 0 (7-20)
The isotherm itself has the form
ey" (721)
Witere a/has a value greater than unity. Owing to ts fexbilty, the Freundlich
isotherm usually fits experimental data aver a reasonable range of concentrations.
For exemple, hydrogen gason tungsten and sulfur dioxide (in aqueous solution)
on activated carbon ** Inthe latte system, the concentration form of Eq, (7-21) at
25°C was found t0 be
Cao: = O180f(Cy)s0,}' * 7-22)
+A. Feamkin and A Slygin, Acta Physice chim. 3,791 (1035) S Brunauer.K.S. Love and R.G.
‘Kozan, J. tn. Chom. So, #4, 751 (1982)
TPH. Emmett aed, Brunaser. J. Ait Chea. Soc. $6, 35 (1934)
[G. Haley and HS, Taylor. J Chom Phys, 18,634 (947),
# RM.W. Teaoel, Proc, Roy Soe (London, A206, 39 (1951)
17 Hiroshi Komnyama and J. M-Smih, AICKE 121, 664 (1973).
su,
oor oa 86 TO
igre 7.8 Heats of edsorpion va solace corerage for hydrogen on metal ls
320 chemical eNamtenise xiveries
where
concentration of SO, in water, g mol/em!
© = adsorbed concentration of SO;. g mol (g of carbon)
In conclusion, note two points. First, the Freundlich isotherm caa be reduced
to cither the Langmuir or the Temkin form by prOper simplication. It may be
‘considered a general empirical form cncompassing the other more specific types.
Second. a single isotherm of the Langmuir or Temkin type cannot be expected to
fit data over the entire range of 0
7-7 Rates of Adsorption
As already noted we are going to use the Langmuir formulation (Eqs. (7-10) and
(7-11) for adsorption rates in Chap. 9 to derive rate expressions for catalytic 1ezc-
tions, Also as noted, the Langmuir formulation is seklom substantiated over @
wide range of adsorbent concentrations, Therefore, iti useful to examine what is
known about adsorption rates and compare this information with the Langmui
postulates
The rate at which molecules of a gus strike a suriace, in motecules (s)em*
surface), is p/(2xmnkpT)!2. Ifs is the fraction of the collisions which resu
chemisorption, that's, the sticking probability. the rate of adsorption is
sn i
Gens Osh
where p is the gas pressure and mis the mass of the molecule. As Hayward and
Trapnell point out,t 5 < | for several reasons, Two ace particularly important:
only those molecules possessing, the required activation energy can be chemi-
sorbed, and molecules possessing the necessary energy may not be chemisorbed
because the configuration of molecule and surface sce may net alow theactivated
complex to be traversed, The fraction of the molecules possessing the required
cnrgy will be e“€*1, where £ is the activation energy for chemisorption. The
configuration probability for a molecule occupying @ single sitet will be pzopor-
tional (o the fraction of the surface that is unoccapied, that is, 1 ~ 0. Thus
all = Be
exr 0.
4)
‘where a is the proportionality constant. often called the condensation coefficient
Combining Eqs. (7-23) and (7-24) yields
Baer -4) (25)
+0... Hayward and 8. MW. Teapnel “Chemizorption” M1 a Butterwasth & Co. (Pobe
Istes) London, 1968
ita molecule (or example. O,)isacates ypon adsoepton and eccpies to adjacent tthe
configuration probably wl be {1 0) at leat fr al sacs of 8.
HETEROGENEOUS PROCESSES, CATALYSIS, AND ADSORPFIDS. 321
‘This ideal expression for the rate of adsorption does not usually agree with
‘experimental data. Observed rates decrease so rapidly with increasing coverage J
‘that they can De explained only i the activation energy increases with 6. Also. the
‘condensation coefficient may vary with (. These variations may be ceused by
surface heterogeneity: that is, the activity of the sites varies, so that different sites
possess different valucs of 2 and £. The mos: active sites would have the lowest
activation energy and would be oscupied first. In addition, interaction forces
‘between occupied and unoccupied sites can cause deviations. In any event, itis
necessary to rewrite Eq, (7.25) showing 2 and E as functions of 0:
ohne
Ont)
‘Comparing the Langmuir equation, Eq, (7-10), with Eq, (7-26) shows that ? in
Eq. (7-10) is a function of surface coverage However, the eeason x and E are
fnctions of @ is that the first two postulates of the Langmuir ueatment (See
|) are not satisfied experimentally; that is, in real surfaces all siter donot
have the same activity, and interactions do ex’.
For many cases of chemisorption the ‘ariation in rate with surfuce coverage
‘an be accounted for entirely in the exponential term of Eq, (7-26), because 2(@)
and 1 —0) are so much weaker functions. This leads to the result (for constant
temperature)
10) 726)
r= Bre
commonly Known as the Elovch equation Equation (7-27) can be obtained:
from Eq, (7-26) by assuming that xis constant and that E a linear function of #
{ean also be derived by supposing that Eis constant but thatthe numb of sites
48 a function of the coverage and of the temperature $
Experimental data for rates of chemisorption are meager.* By pulse-response
measurementst* adsorption rates of hydrogentt on commercial nickel ard cobalt
(on. Kieselguhr) catalysts at temperatures of about 297 K were fourd 19 be
10 x 10"!and 16 x 10° gmoi/(g eatalyst\s) With a similar technique rates of
physwal adsorption of ethane propane and a-butane on silica ge! Rave been
measured at 50°C. The rate constants, (FC). in the concentration range where
the rate was fstorder [C0 in Eq, (7-13}} were: 167 em (s)g silica gel) for
2
1S. Vu Elovch and GM Zhabrova, Zh Fis Khim. 18,1761 (1939), S. Rogiehy and Vs
Zaldovch. ota Pinaewhin, 1 $54 598 (93),
$D/O Hayward and 8M, WTrapnell, “Ceemborpion” 34 ed. Butterworth & Co, (Pub-
Tihs}. London. 1964
SHA Tapiorand NJ Thon.J. dm Chom Sie74, 416 (1982):M JD. Lou, Ch Re 68.267
960)
* For example see Taslr and Thon. Low, ard Hayward and Trapeell for summases of chemi-
socptin rte messorements.
1G, Padberg and JM. Seth, J Catala, 12, 172 (1968). 3. . Advian and JM. Smith J
Catal, 1 57 97%)
4 Actually exchange rates were measured fs deuterium in an atonphere of hydroren so thatthe
cals surice wat saturated wih hadropen
WGP Schneider and JM Sith, ICKE J, U4, 762 (1968
32D CHEMICAL ENGINEERING KINETICS
C:Hy. 255 for C Hy, and about 1500 for n-€,H,. The inerease in rate constant
vith increasing molecular weight is expected since heavier hydrocarbons are more
easily and rapidly adsorbed. Transitionstate theory (presented in See. 2-6 for
homogeneous gas-phase reactions) has been applied to the prediction of the con-
densation coefficient. Lack of precise knowledge about the structure of the ac:
tivated complex on the seid surface hinders these calculations and the evaluation
of numerical values for the rates
Relore we leave the topic of nates itis important to mention the desorption
step. Since desorption is normally an endothermic provess,? it will have a
significant activation eneray (628 Fig. 2-1). Thus desorption will be an activated
process even wien adsorption is not, I AH, is the heat of adsorption on the ste
the activation energics are related by
F=F,— AH, 028)
Rates of chemisorption of hydrogen on & given nickel catalyst were found to be
onactivated (E, ~ 0) bu: the desorption activation energy was 128 keal’g mol.
about the stme magnitude as the heat of adsompiion (13.5 keal g mol]: For
hydrogen adsorption on 2 cobakt Kieselguhr catalyst, the chemisorption also vas
nonactivated with F, = £2 keal(g mol) §
PROBLEMS
TH tna sketch seule 19 Fig 7 show sshematcally the sopeeiraion prey Wr a frstorter
recurs caalte eatin, Coster thee aes: [a ezehon eoutol (6) Cuon eons. anf} an
12 Explain way massirat
Dower temperatures. 4u
retues he glob rile more a aighe empties han at
Jeri reactions, £0 mast ard hearst teistances ae complementary oF e953
os tate?
st ical ut ina om rotor The tem i inather
but ff telesed tha massttam‘e rvstances are portal (a) Weuld neresing tae cUrDLLENC
the gas eopion nea pte estat srlaze increase or decree te elobsl fate?) I hesste f mat
scthertal ant she (ition othermics would sneaing th tu Dalanse intent of decent the
1 Thoce sa growing need to obtain» fetter understanding of reactions moling buclera. Poe
‘timplesmst al sexagedepool plant for puriying municipal wasiovaior ae bacel pon baci al
uti (cuted suze proc), A specal efarisarleo! cto! exigsion shat he ei
rot eal cael the exiation si sarbomaceas sampounds but abo the carbon compound pros
Tis "foe pouth the sasera. ean iatat on conde a sirel-atk rescor operating conte
owsy ats ste and arearsanctompsoats TB Bod const of an aqueous solution ef gears
EA fos chomisrptio procsue appear te he endothermi. s that desorption in thos instanes
sould be exctherme, Sec JN Thomat ant W. 1. Thoms, “Inireduetion to the Priciple:
Fictropenenis Catansis, pt. Academe Poss Inc, New York, (957
2G. Padberg and 1M. Sir. los ct
E16 Adnan a JM Sree Loe ot
HETEROGENEOUS PROCESSES, CATALYSIS AND ADSORPTION. 323,
(thocarhoeaccour mater) which doc nt coetain bacteria The concenation af gins inthe feed!
[sSecand te vclumetrc few rut ite the evcor Q The ture inthe tak conse hacer ins
Conccataion By and the product ara has a How rate equal 1 @. Ths profi mream comains
tetera and lacose fn concetetionsB and § (both im mgiter)
"The twe major ractone may be uriten jin’ highly simplfed mance) allows
1. CHO, + Bg Finer procuets n> 1)
2 C420, 0, mierons diartetes,
Whatsize particles would be novessary ifthe external surface is tobe 100 m* g
G0" m! ke)? The deasity ofthe particles is 20 g’em? (20 x 10” kgm?)
Sout TON The external surface area per unit volume ofa spherical particle of
diameter dis
&
we 6 4,”
Ifthe particle deasity ise», the surface area, per gram of partie’, would be
6
vay
For dj =2 microns (2 x 10"* em)ané py
5,
20 gem?
15 « 10* em? ig (15 x 10° mé Xs)
S,= 1S mg (15x 10? mk)
This is shout the largest surface aren to be expected for nonporous pat~
ticks. Ifa surface of 100 m? g were required, the spherical particles would
havea diameter of
6 6
es x 10-4 em (0.03 10° m:
= sey Or oe 10°* m)
or
4, = 0.03 micron
Particles assmall as this cannot. at present, be produced economically. It may
be noted that the smaller particles found in a fuidized-bed reactor are
revained on 400 mesh screen. which has a sieve opening of 37 microns.
‘The quantitative effete of intraparticle mass and energy transfer on the rate
when a reaction occurs on the interior pore surface ofa catalyst particle is treated
inChap. 11. The methed of predicting their eects requites » geometric model for
the extent and distribution of void spaces within the comslex porous struciure of
soto carutysrs 329
the particle. It would be best to know the size and shape of each void space in
the particle In the absence ofthis information the parameters in the mode! should
be evaluated ‘rom reliable and readily obtainable average properties. In addi
tion to the surface area, three other properties fall nto this cassification: void
volume, the density of the solid material in the particle, ané the distribution of
void volume according to void size (pore-valume distribution). The methods of
‘measurement of these four properties are considered in Sees, $1 to 8-2
8-1 Determination of Surface Area
The standard method for measuring catalyst areas is based on the physical
adsorption ofa gas on the solid surface. Usually the amount of nitrogen adsorbed
‘at equilibrium at the normal boiling point (~ 195.8°C) is measured over a range of
nitrogen pressures below 1 atm. Under these conditions several layers of mole~
‘cules may te adsorbed on top of each other on the surface. The amount adsorbed
‘when one molecular layer is attained must be identified in order to determine the
area. The historical steps in the development of the Brunaver-Emunett-Teller
tmethod' are clearly explained by Emmett-$ There may be some uncertainty as to
whether the values given by this method correspond exacily to the surface area.
Howsver, this is relatively unimportant, since the procedure is standardized ant
the resalis are reproducible. It chould be noted that the surface area so measured
‘may not be the ares effective for catalysis. Only certain parts of the surface, the
lastive ceaters, may be aetive for chemisorption of a reactant, while nitrogen may
be physically adsorbed on much more of the surface, Whes the eatalyst dis
persed on a large-area support only part of the support area may be covered by
‘catalytically active atoms and this ales may be several atoms in depth. Thus, the
fctive atoms may be together in clusters so thatthe eatalytie surface is less than if
the atoms were more completely dispersed or separated. Forexampke a nickel-on.
ieselguhr catalyst was found to have a surlace of 205 m'/g as measured by
nitrogen adsorption.§ To determine the area covered by nickel atoms, hydrogen.
‘was chemisorbed on the catalyst at 25°C. From the amount of hydrogen chemi
sorbed, the surface area of nickel atoms was calculated 10 be about 40 mg. It
‘would be useful to know surface areas for chemisorption of the reactant at reac-
tion conditions. However, this would require measurement of relatively small
amounts of chemisorption at different, and often troublesome, conditions (high
temperature and/or pressure), for each reaction system. In contrast, nitrogen can
be adsorbed easily and rapidly in a routine fashion with standard equipment
In the classical method of determining surface area an all-glass apparatus is
used to measure the volume of gas adsorbed on # sample ofthe solid material.
+5. Bruna, PH Emit and E. Teller. J_ tm, Chem. Sec. 60, 309 (1038)
PLE Emmet (ed) “Catajss” vol. shag 2 Remol! Pcblsting Corporation New York,
ross
'§G Padberg and JM. Sith, J Cay 12 111 (968)
1 Fora complete deveripion ofapparaun snd estioiuca ee LG. Joyner. Siete and Inds
irl Gass Bowlagand Laboratory Techniques Isruments Fubtating Compary.Patsburah 1909;
tee abe §. Bramar "The Adcarption of Gaces and Vagos” vol I, Princeton Universi Pres,
Praxeon 8. 1983,
330 cwneial sveINeRING KINETICS
| 1
we L
Serr |
Aa -1ase|
frost 15"
Figure #1 Adsoroton isotherms for various
sate on 4 0404 sample of slic gel (by
permostonfrm PA, Emma el)" Cotas
fol, Rembold Pablhing Corporation, New
Procure, om York 1954)
“The apparatus operates at 2 low pressure which can be varied from near 2270 up
to about | aim. The operating temperature is in the range of the normal boiling
point. The data obtained are gas volumes ata series of pressures in the adsorption
chamber. The observed volumes are normally corrected to cubie centimeters at
°C and | atm (standard temperature and pressure) and potted against the press-
ture in millimeters, oF as the ratio of the pressure to the vapor pressure al the
‘operating temperature, Typical results from Brurauer and Emmett’s work? are
shown in Fig. 8-1 for the adsorption of several gases on a 0.606-g sample of silica
gel. To simplify the classical experimental procedure a flow method has been
developed in which a mixture of helium (or einer nonadsorbed gas) and the gxs 10
be adsorbed is pasted continuously over the sample of solid The operating total
pressure is constant, and the partial pressure of adsorbable gas is varied by chang-
ing the composition of the mixture. The procedure i¢ to pais a mixture of known,
‘compositionover the sample uncil equilibrium is reeched, that is, uti the solid has
adsorbed an amount of adsorbuble component corresponding to equilibrium at its
partial pressure it the mixture. Then the gas is desorbed by heating the sample while
a stream of pure heliurn flows over it, The amount desorbed is measured with a
‘hermal-conductivity cell or other detector. This gives one point on an isotherm,
such as shown in Fig. 8-1. Then the process is repeated at successively different
‘compositions of the mixture until the whole isotherm is obtained,
P.M, Emits J. dm Chem Son 59. 268 (1937
$M Nebion and FT. Eggeten, Aral Chom, 30,187 (1958)
FA decription ef the operating procedure and the data obtained ate sven by S. Masamure ant
3-M Smith [AICHE J. 10246 (1964) for the adsorption of napen on Vyeor (porous)
SOUD CATALYSTS 331
‘The curves in Fig. €-1 are similar to the extent that at low pressures they rise
rmore or less steeply and then flatten out for a linear section at intermediate
pressures. After earelul analysis of much daa it was concluded tha thelower part,
of the linear region corresponded to complete monomolecular adsorption. If this
point could be located with precision, the volume of one monomolecbar layer of
125, 5, could then be read from the curve and the surface area evaliated. The
Brunauer-Emmett-Teller method locates this point ‘rom an equation obiaincd by
extending the Langmuir isotherm to apply to multilayer adsorption, The
‘development is briefly summarized as follows: Equation (7-12) can be rearranged
to the form
(1)
7 Re tin
Brunauer, Emmett, and Teller adapted this equation for multilayer adsorption
and arrived at the result
WRo= A) tne * inp
where py isthe saturation or vapor peesure and ¢ isa constant forthe particular
temperature and gas-solid system.
‘According to Eq, (8-2) plot of pe{y — p) vs: p/po should give a steaight line
The data of Fig. 8-1 are replotted in this fashion in Fig. 8-2. OF additional
ignifcance is the fact that such straight lines ean be accurately extrapolated to
Pat Ce (62)
cane
(Data based uvon 0.606
‘ample of sia ge.)
The otdinute for curve 7
| thould be muttinhot by 16
. BI OF B51 ~ cues 1
© 02 04 = 08 OR ~ Curve?
vm
[igure $2 Plot of Brunauer-Emme-Teller equailon (Bq, (8-2)] fr data of Fig. 8 [by permasion
‘from PH. Enmat (et * Catalysis to. Relshld Publishing Corporation, New York, 1934
232 enicat BNarSEEANNG KINETICS
pipe = 0. The inteceept 1 obtained from this extrapolation. along with the slope s
Of the straight line, gives 10 equations from which ©, can be obiained,
at pip 0 3)
(8-4)
Solving these equations for the volume of adsorbed gas corresponding 10 a
monomolecular layer gives
Its Be
“The volume v, can be readily converted tothe number of molecules adsorbed
However, to deternine the surage arcs it's necessary to sccet a valuofor the aca
covered by one adsorbed molecule I tis i 2 the total surface area is gwven by
cg fs 6)
where Np is Avogadro's number, 602 x 19"? molecutes/mol, and V isthe volume
pet mole of gas al conditions oft. Since ry is recorded at standard temperature
ar’ pressure, V = 22.400 cm?g mol. The term in brackets represents the number
‘of molzeules adsorbed. Ife, is based on a 10 g sample, then S, i the total surface
per gram of solid adsorbent
Emmett and Brunauer* proposed that 3s the projected area ofa molecule on
the surface when the molecules are arranged an close two-dimensional packing
‘This value is slighily larger than that obtained by assuming thet the adsorbed
molecules are spherical and their projected area on the surlac> is circular. The
proposed equation is
(87)
where Mf is molecular weight and p is the density of the adsorbed molecules.
‘The term in brackets repiesents the volume of one adsorbed molecule, The deasity
's normally taken as that of the pure liquid at the temperature of the adsorption
‘experiment. For example, for Ny at ~ 195.8°C, p = 0.808 gem! and the area per
molecule fram Eq. (8-7) is 162 x 10° em?, or 162 A? Hi this result is used in
Eq, (8-6), along with the known values of Ng and V, the surface area pet gram
S,=435 x 10%, om? g solid adsorbent (88)
Table 8-1 shows surface areas determined by the Brunauer-Emmett-Teller
method for a number of common catalysts apd carriers. Calculations of surface
areas from adsorption data ate illustrated in Examples 8-2 and 8-3.
+ PLHL Emmatand'S Bruniuer J son Cho Sac. $9, 1553 (197)
socio CATALYSTS 333,
‘Table 8-1 Surface arca, pore volume, and mean pore radii for typical
solid catalysts
Swifecr aren Pure volume
Catalyst mg enig Mean ere radius, A
Auivacd item S015 0608
Sica gs rw os
‘SiQnAl,O, cracking
“atalyie 0050p 02.07
Acated lays isos 04.032
Activated alumina 175 033
Ceite (Kinselguve) 42 i
Syathetic ammonia
aati, Fe on 20-190
Pumice ox
Fed copper 023
‘Source: Impact kom A. Whew, “Advance in Catal
Academic Pres, The, New Yor, 1950,
pp 20-326.
Esample 82 From the Brunauer-Emmett-Teller plot in Fig. 82 estimate the
surface area per gram of the silica gel. Use thedata for adsorption of nitrogen
at ~ 195°C.
Sonution From curve 6 of Fig. 8-2, the interoept on the ordinate is
OL x 10-3 em=?
The slope of the curve is
GIA ON KIO" os eiysee®
a
These values of s and J may be substituted in Eg. (8-5) 10 obtain ty,
wo
Pa gr iy ag 7 BSM/EcHabst
‘The factor 0.606 is introduced because the data in Fig. 8-2 are for asilica
gel sample of 0606 g, and is the monomolecular volume per gram. For
nitrogen at —1958°C, the application of Fa, (8-8) yields :
S, = 4.35(126) = 550 mg
Example 8-3 For comparison, estimate the surface area of the silica gel by
using the adsorption data for oxygen at ~ 132°C. The density ofthe liquefied
‘oxygen at — 183°C, from the Intemational Critical Tables, is 1.14 g em.
Soucriow First the area of an adsorbed molecule of ©, must be calculated
from Eq. (8-7):
2
oof 142. x 10° cm®
* [gars 10" al (een
{M4 CHRWICAL ENGINEERING KINETICS
With this value of 2 Eq, (8-6) gives
(602 X10) 445 yg-r0
a2 x 10
From curve 4 of Fig. (8-2),
1 = 0.40% 10°) m3
tor = 132% 107) em->
Bx 10%, emg
‘Then the monomolecular volume per gram of silica gel is from Eq. (8-5).
ee
o.4 + 132 (0608)
Finally, substituting this valve of fq in the atea expression
Sp= 38 x 1O*(122) = 465 x 10" em*ie oF 65 mg
The difference in area determined from the N. and O, data is somewhat
larger than expected fr these gases. The adsorption curve for Ny at — 183°C
ives a value in eloser agreement with $50 m?ig (se Prob. 8-4}
= 122 em! g catalyst
8-2 Void Volume and Solid Density
The void volume. o pore volume’ of a catalyst particle can be estimated by
boiling a weighed sample immersed in a liquid such as water. After the air in the
pores has been displaced, the sample is superficially dried and weighed. The
increase in weight divided by the density of the liquid gives the pore volume.
A mote accurate procedure the Jeliun-mercury method. The volume of
thelium displaced by a sample of catalyst is measured; then the helium is removed.
and the volume of mercury that is displaced is measured, Since mexeary will not
fil the pores of most catalysts at atmospheric pressure. the difference in volumes
ives the pore volume of the catalyst sample. The volume of helium displaced isa
‘measure of the volume occupied by the Solid material. From this ard the weight of
the sample. the density of the solid phase, 2s. can be obtained. Then the void
fraction, oF porosity, of the particle, ¢,. may Be calculated from the equation
void (pote) volume of particle cAA
"total volume of particle ~ mV, + mV pa)
Ys
= 9)
Yas +1 bas
where ic the mast of the particteand tithe void volume per gram of pati,
Tithe sample of parce weighed, the ass divided bythe mercury volume svt
the density p, of the porous particles. Note that the porosity is also obtainable
from ths density By the expesion
y,
ved oli 7
“+ iotal volume = Tip, 7 Pet ©)
sip oxrauver 335
From the helium-mercury measurements the pore volume, the solid density,
and the porosity of the catalyst particle can be determined. Values of: are ofthe
‘order of 0.5, indicating that the particle is about half void space and half solid
‘material. Sinee overall void fractions in packed beds are about 04, a rule of thumb
for a packed-bed catalytic reactor i that about 30%, of the volume 1s pore spa
10% is solid catalyst and carrier, and 40°, is void space between catalyst particss|
Individual catalysts may show results considerably difleent from these average
‘values, as indicated in Examples 8-4 and 8-5 and Table §-2.
Example 84 In an experiment 10 determine the pore volume and catalyst
particle porosity the following data were obtained on a sample of activated
silica (granular, 4 to 12 mesh size)
Mass of catalyst sample placed in chamber = 1015 g
Volume of helium displaced by sample = 45.1 cm?
Volume of mercury displaced by sample = 82.7 cm*
Calculate the required properties
Sotution The volume of mercury displaced, minus the keliun-displacement
volume, i the pore volume. Hence
BI- 451
115
‘The helium volume is also a measure ofthe density of the sold material in the
‘catalyst; that is,
Y= = 0371 an%ig
ois >
bem Fey 2228 glo
Substituting the values of ¥, and 9s in Ea. (8-9) gives the porosity ofthe silica
gel particles,
0371(225)
© = ape OS
Catalyst particles used in packed-bed reactors normally are pelleted of ag-
slomerated by other means, to sizes of fin. to din. in order to avoid excessive
Dressure drop and to provide mecharical strength, Usually the pellets arecylindi-
tal or granular, Agglomeration of porous particles gives a pellet containing 10
Void regions: small void spaces within the individual particks end larger spaces
between particles, Hence auch materials are suid te ceniain bidisperse. pore
systems, Although the shape and nature ofthese two void regions may vary from
thin eracks to a continuous rogion surrounding ¢ group of particles, it has been
customary to designate both regions as pores. The vord spaces within the particles
‘are commonly termed micropores, and the void zegions between parcicles are
called macropores, Particle refers only to the smal! individual unit irom which the
pallet i produced
Pethaps the most widely used pellets are those of alumina, Porous alumina
particles (20 10 200 microns diameter) containing micropores of 10 to 200 4
336 CHEMICAL ENGINEERING KINETICS
Table 8.2 Physical properties of alumina pellets
Density pam? Pore volume. cm? § Void frastion
Panicle Pelt Mico Macro Toul Maco Mire
iL OMs mos CIM cae
126 10100319} OT
' 08% 040 0305 Oaks 889
122 GTS 0416 SL 580383 CT
Vise © 062 out = sm = 72s Gas. aRNs
avs: All properties ate based on ALO, Micro refers to pore radi le than
180 A.and macte refers to ead greater than 700 A
Source RA. Micke a |. M.Smith, fd Eng. Chm Fond Quart,
(962)
diameter are readily prepared by spray drying. These somewhat soit particles are
easily made into pellets. The mucroporosity and macropore diameter depend on
the pelleting pressure and can be varied over a wide range, Table £2 shows macro
and micro properties of five alumina pellets, each prepared at adifferentpelleting
pressure, The pellet density listed in the second column is approximately peopor-
tional (0 the pressure used. Comparison of the least and greatest pellet density
shows that the macropore volume has decreased from 0.670 to 0.120 with ine
crrasce pelietaig pressure, while the micropoxe volume has decreased only from
0434 to 0.365,
‘The external surface area of even very fine particles has been shown
(Example 8-1) to be small with respect to the internal surface ofthe potes. Hence,
in a catalyst pellet the surface resides predominantly in the small pores within the
particles. The eaternal surface of the particles, and of course the external area of
the pelles. is negligible
Macro- and micropore volumes and porosities for bidisperse catalyst pellets
are calculated by the same methods as used for monodisperse pore systems.
Example 8-5 illusirates the procedure
Exanyle 23/0 yrogeton saat regaret by senting lisa pr
ticles (100 to 150 mesh size) in aqueous NiNOy solution. After drying and
Tesla tc abi Gace Oa Tats ee eae
Beirne cheese euler puoeersiree
a
Sas. wile;
Beean tak
Pass =1e
Volume = 3.2m"
‘The AIO particles contain micropores, and the pelleting process introduces
macropores surrounding the particles. From the caperimental methods
already described. the macropore volume of the pellet is 0.645 em? and the
micropore volume is 040 cm? of particles. From this information calculate:
souD CATALYSTS 337
(a) The density ofthe peiet
(b) The macropore volume in cubic centimeters per grarm
(c) The macropore void fraction inthe pellet
(d) The micropore void fraction in the pellet
(€) The solid fraction
(J) The density of the particles
(9) The density of the solid phase
(i) The void traction of the particles
SoLurION
(a) The density of the pellet i»
aus
rem a
(b) ‘The maeropore volume per gram is
06s
(n= SSS = 0208 em'/g
= 0978 jem?
(c) The macropore void fraction cy is obtained by applying Eq, (8-10) 10 the
pellet. Thus.
macropore volume _ (Vi)w
ow = ial volume = Vp
(d) Since
(H),= 040 om'/g
the micropore void fraction », in the pellet is
Ue 040
Upp ~ 10978
(€) The solids fraction e, is given by
0391
Vatut tte
25= 10200 0.391 ~ 0.409
() The density p, of the particles can be calculated by correcting the total
volume of the pellet for the macropore vokime, Thus,
0978
me jean
o20s(oo7R) ~ 1
23 cHICAL ENOINEERING KISERICE
(9) ‘The density of the solid phase is
mass of pellet,
Wolume of pellet) és
be 0978
a 09
bs
230 giem!
li) The void fraction of the particles is given by
.
7 fa = Pole
(0.40) = 049
For this pellet a fraction equal to ny +5, = 0.591 is void and 0.409 is
solid. OF the individual particles. a fraction 0.49 is void. Note that all these
results were calculated from the mass and volume of the pellet and the
‘measurements of macro and micropore volumes,
8:3 Pore-Volume Distribution
We shall soe in Chap, 11 that the effectiveness of the int
reaction® :an depend not only on the volume of the void spaces (14). but also on the
radius of the Void regions. Therefore its desirable 10 know the distribution of said
volume in a catalyst according to size of the pore. This is a dificult problem
because the void spaces in a given particle are nonuniform iu size. shape. and
Fength. and normally are interconnected. Further, these characteristics can change
fcom one type of catalyst pattisle to another. Figure 8-3 shows elsctron-micro-
scope (scanning type) photographs of porous silver particles (S, = 197 m*'g). The
‘material was prepared by reducing a precipitate of silver fumarate by heating at
350°C in a stream of nitrogen. The larger darker regiors probably represent soid
space between individual particles. and the smaller dark spaces are intraparticle
voids. The light portions are solid silver. The complex and random geometry
shows that itis not realisti¢ to deseribe the void spaces as pores, It's anticipated
that other highly porous materials such as alursina and silica would have similar
continuous and complex void phascs. For a material such as Vycor, with its
relatively low porosity (0.3) and continuous solid phase, the concept of void
spaces as pores is more reasonable.
In view of evidence such as that in Fig. $.2. it is unlikely that detailed quanti
lative descriptions of the soid structure of solid catalysts will become available
Therefore, to account quantitatively for the variations in rate of reaction with
location within a porous catalyst particle. a simplified model of the pore structure
's necessary. The model must be such that diffusion rates of reactants through the
Void spaces inio the interior surface can be evaluated. More is said about these
models in Chap. 11. It is sufficient here to nots that in all the widely used models
the void spaces are simulated as cylindrical pores. Hence the sizeof the void space
's interpreted as « radius a of @ cylindrical pore, and the distribution of toic
volume is defined in terms of this variable. However. as the example of the silver
SoU CATALYSTS 339
rograph of potoussiver partie appronimate surface area = 19.7
39000): megefeaion = 1K (ter = 10.00 A)
3M comics: eNerserane eves
catalyst indicates, this docs aot mean that the void spaces are welldefined eylin=
drical pores
There are two established methods for measuring the distribution of pore
volumes. Themercury-penstration method depends on the fat that mercury haw a
ficant surface cension and does not wet most catalytic surfaces. This means
that the pressure required to force mercury into the porss depends on the pote
fadity The pressure varies inversely with a: LODIbiin# (approximately) &
Fequired 0 fill pores for which a = 10,000 A. and 10,000 Ibvin is necded for
4= 100 A, Simple techniques and euipment are satstactory for evaluating the
pore-solime distribetion down to 100 10 200 A, bat special high-pressute appar-
tus is necessary 10 g0 below a = 10D A, where much of the surface resides Lathe
sevond method, the nitrogen-adsorption experiment (described in Sec. 8-1 for
surface area measurement is continued until the nitrogen pressure approaches the
Saturation value (1 atm at the normal boiling point). At p py — 1.0, where py B the
saturation pressure, all che void volume it filled with adsorbed and condensed
nitrogen. Then « desorption isotherm is established by lowering the pressure ia
increments and measuring the amount of nitrogen evaporated and desorbed for
cach increment. Sine: the vapor pressure ofa liquid evaporating from a capillary
enends on the radius of the capillary, these date can be plotied as volu
ovorbed vs, pore radius. Thus this procedure ae gives the distribution of pore
Slims The vapor pressure i net alected significantly by radii of curvature
uisaic: than about 200 A. Hence, this method i not suitable for pores larger than
San A
Ia high-pressure mercury parosimeter not available a combination of the
‘s49 methods is necessary to cover the entire range of pore tai: (10 10 10.000 A)
‘hich may est ina Did sperse catalyst or support. such as alumina pellets Fer 2
‘monodperse pore distribution, such as that in silica gel the nitroger-desorpion
experiment is sufficient. since there ae few pores of radius greater that 200 A In a
bigisperse pore system the predominant part of the cuialytie eaciton occurs in
Fores less than about 200 4 (the micropors region). since that s where the bulk of
turfacs resis. However the tansport of Feactanis to these small pores occurs
primastly in pores of 20 to 10.000 & (the macropoce region) Hence the complete
Sistrbution of pore volume s required inorder to establish the ellctiveness ofthe
Interior surlace. that is. che global rate of reaction, Calculation provedures and
{ypical results are discussed briefly inthe following paragrapns.
Mercury-penetration method By equating the force due ta surface tension (which
tends (0 keep mercury out of a pore) to the applied force, Ritter and Drake
obiaines
= -2rae eos 0
(ey
FHL Rien LC Desks, Pa Bay Chen dnl 19,787 (1948)
soup caranysts M1
Figure 4 Mercury peaetation in a pore of radius
whore (is the contact angle between the mercury and pore wall (Fig. 8-4), While 0
probably varies somewhat with the nature of the solid surface, 144” appears io be
‘a good average value. Then the working equation for evaluating the radius corre:
sponding to a given pressure is
8.75 x 10°
a@(A) (S12)
(= (bin) y
Caleulation of the poresize dstribution by th method illustrated in
Example 8-6,
Example $6 The mercury-penetration data given in Table 8-3 were obtained
on a C624 semple of a uranium dioxide pellet formed by sintering
particles at 1000°C for 2 h. Since the particles were nonporous, the void
space was entirely between the particles (that is, in macropores), At the
beginning of the experiment (when the pressure was 1.77 Ibyin.* abs) the
amount of mercury displaced by the sample was found to be 0,190 cm’.
Calculate the poresity and pore-rclume distribution of the pellet.
Sovurion According to Eg, (8-12) at p= 1.77 Ib/in? abs only pores larger
than about $00000 A (50 microns) would be fied with mercury. No pores
larger than this are likely. Hence 0.190 em? isthe total volume ofthe sample,
At the highest pressure, 3.500 Ib/in® abs, only pores less than a= 250 A
‘would remain unflled. Since the pores were entirely ofthe macro type, ke”
poressmaller than 250 A ae expected. Neglecting these pores, the porosis can
bbe calculated from the porosimeter measurements alone, Thus
0.125
*= 9190,
66
(M2 crtsucat exanveentve amertes
‘Table 8-3 Mercury porosimeter data for
uranium dioxide pellet (mass of sample
0.624 2)
Mery
Presse concetation ron,
ee! cm
ne owe 0196
30 B06 Ds
ue ous
yao bam
S10 p00 0167
36006 bis?
et poe oust
SOs ou
050 one
moO 00?
mat oer
se Day 971
0 Gos 0st
oso 003
1300 on20
so ore
90 boo
22 00)
io °
A check on this result is available from air-pycnometer* data, which gave a
solid volume of 0.0565 em?. Thus the total porosity is
0.190 - 00565
= Ti90 —
The dference between values suggests that there were a few pores smaller
than 250 A inthe sample although the comparison also includes experimen-
tal errors in the t4o methods
To calculate the pore-volume distribution the penetration dasa are first
corrected 10 a basis of I g of sample, as given in the third columa of
Table 8-3. If we neglect the pores smaller than 280 A. the penetsation data can
be reversed, starting with V = 0 at 3500 Ib ia abs (250/A). The last columa
shows the figures. Then, ffom Ea. (8-12) and the pressue. te radius corre.
sponding te each penetration valus can be sstablshed, This gives the penets:
lion curve shown in Fig. 85. The penetration volume at any pore radius @'8
the volume of pores larger than w. The detivative ofthis curve. QV a. the
volume of pores between a and « ~ Aa divided by Aa:that is, tthe distribu
sion function forthe pore volume aceosding to pore radius. tis customary 19
n
hich vce siratewo prenunerte mesure vol valu of puro ete I prods
asthe Reta measurement desebal Ses 82
soci0 caratvers 9
— on
ot 4 1035
“rtf xe
us| 7 aS
on} —t—} 7 d on
008 | oat as =
Dursabeane |S
0s ral Ty
ons aos
[7 Peneiration curve
Lo Lal are J
ree 3 656 8 12 3 456 8
Pore radius 4 A
Figire 85 Porevolame dictibation in «UO, ple
plot the pore radius on a logarithmic coordinate as shown, Hence the distri-
bution function is taken as the derivative of the curve so plotted, that is,
Vid (log a) The distribution furction is also shown plotied against a in
Fig $5,
For this pelles the distribution is seen to he reasonably symmetrical with
‘most of the volume in pores from 300 (0 8000 4 and with a most probable
pore radius of 1200 A, Note that the flatness of t=e penetration curve at low
pore radii justifies neglecting the poces smaller 1:20 250 A,
Wheeler? has summarized the assumptions and accuracy of the mercury-
penetration method. ft is important to nete'that erroneous results would be
obtained if the porous particle contains large void spaces thal are connected only
to smaller void spaces. Suc large “bottlencek ” pores would fll with mereury et
the higher pressure corresponding to the connecting smaller pores. For accurate
resulis eachporous region must be connected to at lsast one larger pore.
‘Nitrogendesorption method As the low-temperature nitrogen-adsorption experi-
‘ment (Sec, 8-1) is continued to higher prestures multilayer adsorption occurs, and
ultimately the adsorbed films are thick enough co bridge the pore-t Then further
uptake of nitrogen. will esult ia capillary condensation. Since the vapor pressure
‘dzcreases as the capillary sizz decreases, such condezs.tion will oceur first in the
smaller pores. Condensation will be complete, as pp. ~ 1.0, when the entire void
1 Acne, PHL mint ed, Calais” vo 1 p32
New York, BSS,
ELH Cohan J. Am Chon, Soc, 60133
way
1 Fusing Corporation,
{088} AG. Feces J. Phas Coll Chom. 88.638
MA chrtea ENGINEERING KINETICS
region is Filled wth condensed nitrogen. Now, ifthe pressure is reduced by asm
increment, a small amount of nitrogen will evaporate from the meniscus formed at
the ends of the largest pores. Pores which are emptiad of condensate inthis way
will be those in which the vapor pressure of nitrogen is greater than the chosen
pressure. The Kelvin equation gives the relationship between vapor pressure and
fadius of the concave surface of the meniscus of the liquid. Since some of the
nitrogen is adsorbed on the surface. and therefore not present because of capillary
‘condensation, the Kelvin relationship must be corrected forthe thickness 6 ofthe
adsorbed layers. With this correction, the pore radius is related to the saturation
pressure ratio (vapor pressure pin the pore divided by the normal vapor pressure
ro) by
= a 20h, 008
RT In wip)
where Hj = molal volume of the condensed liquid
= surface tension
= contact angle between surface and condensate
-6
(6-13)
Since nitrogen completely wes the sucface covered with adsorbed niteogen.0-= 0°
and cos = I. The thickness 6 deperds on p/pp. The exact relationship hes been
the enbject of considerable study.* but Wheeler's form
log) oy
is generally used.
For aitrogen at —1958°C (normal boiling point) Eq. (8-13), for a
angstroms, becomes
*
silos)" eas
with § determined from Eq, (8-14)
For a chosen value of p/po. Eqs. (8-15) and (8-14) give the pore radius aboxe
which all pores will be empty of capillary condensate. Hence, ifthe amount of
desorption is measured for various p/p the pore volume corresponding t0 vari-
fous radii can be evalvaied. Difleentiation of the curve for cumulative pore
volume vs. radius gives the distribution of volume as described in Example 8-6.
Descriptions of the method of computation are given by several insestigators.£ As
in the mercury-penetration methed, errors will result unless each pore is Con
nected to at least one larger por.
Figure £.6 shows the result of applying the method to a sample of Vycor
+A. Wheeler, in P, H: Eman (o.) “Catala” vol. chap 2, Renkol! P2bishing Cerpors
tion, New York. 1959;6, D. Hakey J. Chem Phe. 16,931 (1988):C. GS, 4m. Chom. S07
1105 (948): 5. © Mingle aed 1M. Sma Chom En. Sei. 16,31 (1961
LEP. Barrett L.G Joynsr. and PP Halen, J. Am. Chom. Soc. 78,393 (1961); €.3. Pre J
Prys. Chem, 9, 109 1959), B Anderson, J Catal 3, 501864)
sou CATALYSIS 345
T
" L |
‘ rh
res |
:
z
re
|
-++—-F
ary 2S ee ie
reves A
‘Meare 46 Foxe rohume dstitution la Vseor: 9, © 86 gem?,, = 0208 omg §5=90 mg
vorous glass).+ This materia, which contained only micropores, had the
properties
0, 146 glen?
¥,= 0208 om? 'g
= 0305
= 90 mig
‘The surface area was determined from eitrogen-ndsorpiion data io the 1o¥ ppp
mgs. as described in Sec 8-1, while the distribution resus in Fig. 86 were
‘stablished from the desorption curve in the sapillary-condensation (high p Po)
region
By combining mercury-penetration and niteogen-desozption measurements,
Pore-rolume information can: be obtained Over the complete range of radi in a
pelleted catalyst enctaining both maceo- and micropores. Figure §-7 shows the
omutative pore volume fr two alumina pullets, each prepared by compressing
‘orous particles of boehmite (A13O,:H,0} Ths propectiess of tne two pellets are
given in Table 8:4. Th only difernce in he two isthe pellstng pressure, Increat-
lng this pressure causes drastic ceductions in the space between particles (maceo>
ore volume) but does not srealy change the void volirne within the particle: or
M, R. Rao and 1.M¢ Sith AICHE J. 10.599 (969)
‘Tee propetio and porevoluns dicribuien were delemiced by M. F.L Johnsoa. Siccis
oath Laboratories. Harvey. by We melbodsdseibed it Sc. 1 te &3 They wate orinaly
repost mL. Roterisow and JME Smith AICHE J. 34 (198),
g
HEICAL ENOINEERSG RIMES
:
a HH
: root
Pore radon a. A
gate #7 Pose volume in aemins (Boehm) pets
the surface acca, The derivative ofthe volume curves in Fig. 8-7 gives the pors-
volume distribution, and these results are shown in Fig. && ln this figure the
bidisperse pore system, characteristic of alumina pelicis. is leary indicated. The
rmicropore range within the particks is narrow. with a most probable radivs of|
20A. The macropores cover a much wider range of cadit and show the effect of
pelleting pressure. For the high-pressure pellet ll yolume with pores greater than
2000 A has beco squeezed out, while the most probable radius for the tow
Dressure pellet is S000 A, Pelkting pressure seams to have litle effet on tie
micropores, which suggests hat the particles themielves are not crashed
Significantly ducing the pellting prooss.
Some models (i Chap. 11) for quantitative treatment ofthe elfectivenes of
te internal catalyst surface requite only the average poceradivsd.cathse than the
distribution of pore volumes. Wheclert has developed a simple equation for a
1A. Wheto, nM, Erne fet “Cua” vb. chap.
Roots Pobishing Corpor
‘oan, Now York. 1955.
“Table 8-4 Properties of bochmite (41,0 5*H,0) pellets
Potating_Macropore volume. Misoporsvoluns, Surge arc,
Presare cm! s ome. ae
tow toe 086. 7»
ah 38 oe a
Meter: Volume vod sfce ste ree 19 mae of A,O, obtained by
‘lon of bochmits, The pocevume digrietion fs wven in Fig. 8, Average
‘pate tze rom whch pelts wers made was BS irons,
‘sou cxtatysts 347
E Low netleung pressure |
TP 2 High pelleting preesore"| 7
Wag a sis
Py OR 234 OST 2 I+ OS ZL GEM
Pore radios. A
Flew #8 Pove-vetunsditshution in shins palit
which requires ooly surface-arca and pore-volume measurements, Suppose all the
‘pores in a hypothetical particle are steaight. cylindrical. not interconnected, and
hhave the same cadius d and length £, The average pore radizs may be found by
weiting equations for the total sarface and volume in the hypothetical partick= and
equating these quantities to the surfuce mS, and volume mF; in the actual
particle: ie
1,5, (240L)n 16)
60)
where m, and # are the mass and rumber of porcs per particle Dividing the 10
equations gives the average pore radius
2"
ante ts
3 (sts)
‘This expression agrees well with volume-average values obtained from the
distribution curve for monodisperse pore systems. For example, om the dats for
the Vycor sample (Fig. 8-6) Eq. (818) gives
210208)
x10"
S46 x10 om or do A 4
‘The volume.average Value is calculated from the porevolume data used to obtain
the distribution curve (Fig. 8-6) and the expression
Ly this method d= 43 A.
Accurate valucs of the small areas existing in macropore systems make it
dificult co use Eq. (8-18) (0 calculated for interparticle porcs. Hence the average
radius for systems such as the UO; pellets discussed in Example 8.6 should be
biained by integrating under the cumulativesctume-vs.
reactant molecules,
Selectivity poisons ‘The selectivity of a solid surface for catalyzing one reaction
With respect 10 another is not well understood. However. it is known that some
materials in the reactant siceam will adsorb on the surface and then eataly ze othe:
undesirable reactions, thus lowering the selectivity. The very small quantities of
nickel, vanadium. iron, etc.. in petroleum stocks may act as poisons in this way.
‘When such stocks are cracked, the metals deposit on the catalyst and act &s
dehydrogenation catalysts. This results in increased yields of hydrogen and cok=
ard lower yields of gasoline
Stability poisons When water vapor is present in the sulfur diovide-air mixtuse
supplied to a platinur-alumina catalyst, a decrease in oxidation activity occurs
This type of poisoning is duc to the effec of water on the structure of the alumira
RH. Grifch, “The Mechanism of Contiet Catabsis” p. $3. Oxlord Uninerity Press. Xe
‘York, 1936: B Mant. J. Soe. Chem. I Lenn) 67.99 (1588) PH. Emmet
‘ol I, chap. Reithold Publahing Cocporation New York, 1934, Aa teste review by JB. Bs
(Chemical Reaction Engineering. Ady. Chemin Series, 108 259(1972). BW. Wossechowst. C=
Re 9.79 (974),
"Typical rterences are M. Sagara, S. Masamune and J. M. Smith, AICRE J. 13.1226 96
GF Froment and K. 8 Bicholl Chem Eag.Sc16 189 (91): P.B. Weisz and R. D Goods.
Coty 2 971963}
356 cMEUICAL ENeINEERING KNETCS
Table 8-6 Poisons for variows catalysts
Type of
cays escon poscning Poisons
Sica, slemina Cracking Chenisorpion Crgaric bates
Depesiion Carbon
subty Water
Sekcinty Heavy metals
Neel. platinm, Hydrogenation Choniompton Compounds ofS. Se,
copper Denydrogenaton TeP.AL ZA hades,
Hg. Po, NH. CoH,
HS, FeO).
obae Mydrocracking Chemborption NH). 5.50, Fe P
iver CH, +0=C)H,0 Seleainny
Venidium oxide Oridaton Chemisoepion
Irom Ammoria syatheis — Cheninorption
Hyarogeeation Chenisorpioe
Ondator Chenisorption
Source: tm par ftom W. B. ines eH. Eee fe. CaIays8 WoL ena 7. p36,
Reinhold Publishing Corporation New York, 19,
ca:rier. Temperature has a pronounced effect on stability poisoning. Sintering and
Jocalized melting may occur as the temperature is increased, and this, of course,
changes the catalyst structure.
Diffusion poisons This kind of poisoning has already been mentioned in connec-
tign with carton deposition on cracking catalysts, Blocking the pore mouths
prevents the reactants from diflusing ino the inner surface. Entrained solids in the
reactants, or fluids which can react with the catalyst 1o form a solid residuc. can
‘cause this type of poisoning.
‘Tables 8-5 and 8-6 list poisons for various catalysts and reactions. The mate-
rials that are added to reactant reams 10 improve the performance of a catalyst
are called accelerators. They are the counterparts of poisons. For example, steam
added 10 the butene feed of a dehydrogenation reacior appeared to reduce the
amount of coke formed and increase the yield of butadiene. The catalyst in this
cease was iron?
PROBLEMS
1 The sea gel for mhexane adsoration mentiotad ia Prob. 14 has the following propecis
Sp=832 mt ty = 045. p,= LID gem’. Yy— Odom? g. Using the adeorption data gies in
Prob. 1-6 ta) Estimate te fection ofthe sure covered vib an adsorted moromclectat best
eachptil pressure mhecane, The surance cecpied by one molecule of erate at 70°C is estimated
to be 585% 10°" em?; (5) Coeulate the value of C. from the total surface aces andthe ae
+ KK, Keay Ins Erp. Cher 42.295 (1980)
soup carats 387
ovcapiod by one molete of texans: What sonsions cam be draws (oun the eomparion of hi
Gilee of C, wih that obtained in Prob. 782
1B2 Repeal Prob. 8:1 fr benzene aderption oa thesame lea gel but at 10°C. The ssf conupiok
fh one benzene molcrule w estimated to Be 348 x 10" '* cm? Use th adsorption Sats fr benzene
[Been in Prob 77,
3 Rete to Pod. 7-12 Usinga constant value of 48x 10°" efor the wifwe area accep by
tne mokoule of benzene. Getetine the ractoeal surfice coverage at each prtal pessurcor benzene
renin Prod 712,
SeCune Jot Fig. 82) a Brunaver EmmetTeller plot bee adsorpien daa of; at ~189°C 08
thecample fies ge The deity of gud N, atts temperature e731 gem? Eimaerte ate of
Uh ain gel ora these data uate ste per pam, and compare with the rss of Example 8
465 The“poies-B method” of estimating surace areas eas requ ased prot othe development of
the Brenaver Emmett Taller approach. entled choosing fom an acsorplion diagram sich 35
Fig Br the point at whith the central Tine section begin. This proseduce worked sel for some
{ystems butt waseatemey eiffel. sft impossible 0 sees arciabe point Dow an sherm such
ethat stows lor nbutane Fig #1, Incontad, the Brurauer Emmet-Teller nathod was fourd 10
Tereabnanty setacter fr the pe of other, Demonerate this by estimating the sure ara of
the slica get aig from the bene curve in Fig. 8 flip the ordinate ofthe butane cone
Dy 10) The density of gud burae a O'C is 0901 gle"
$6 An Sol gsuple of Glavcouiis sedis wits Nz adsorption at —1958°C. Thefotowing data ae
tained:
Peswgnmiz 6 «250-2 S DOSS
Volume showed 6) 7 MDT OS OOS
cm? (ah °C
apd tem)
“Thovaper prsuts of Na: =198H°Cie Late. Estimate the suracearea (are meters er eramdot
the aie
£87 Low-temperature (—1984°C) nitrogen adserrtion data were obiained fr a9 Fe-A1,0 ,ammoxia
etait Theres for 4 $0.6 sampie were
Pressures meg #3050 10213 148 233 SE SID AD HW HT 50
WO) 116 3) 8 157 9 8S BS
2 2H 56
sd Cat)
Estimate the surface aves fr ths catalyst.
188 fetter and Drake give the te demiy of che old mater in an activated slemina partie at
3675 glam? The deasay othe parte: cetermined ty metearydipluceent 1547, Theowrfase alee
ip alwortion mensurmert s 1754/2 From ths (norratos compute the pre volume pet gram
I poresiy ofthe petcea and the san pore rads. The bulk density of » bed of the alsmina
puis in a250-n? graduates DAT gem" What Faction9' the total volume bed is 0 pace
Eerwecm the particles and what fraction is void space within the partic?
{89 Two samples ofsicrstunlaa cracking catalyst have parle denis of 1136 and 096° gen?
epectnelnasSetermined b) mercury displacement Thettue density f the asl teal nach ete
+H. Rien and EC, Drake, Ind. Eng. Ch, Ant 17,187 (1943)
388 CHEMICAL ENGINEERING KINETICS
15 237 gem? The surace area othe Bist sample i467 big and that of the stsond is 372 mie,
‘Which sume has the larger mean pore radia?
10 Mercury gorosimeer data are buted below for 2 0.400-g sample of UO, pellet. At the
beginning of the measurements [p= L17Ibin? ate) the mercury deplaced by the sample wae
12S em’, Athi low prwure no pores were peacfated.Outa obtained wil « pyenomeicr gave
teve density ofthe solid phase of y= 757 Ro
Calculate the eal porosity othe pale ad the poresiy due to pores oflargerthan 250 Aradias
Ao plot the porevelumedisfutin for she pores age than 280 A radius, using he coordinates of
Fig. 85.
19629586 S10 GODT 900.
Mec 00: OBO Gome Bol4 062 0026 OO 008
{000 1200 400 wen 2840 2900 3300
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5806)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (589)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Assignment 3Document4 pagesAssignment 3Sandeep ChallaNo ratings yet
- Handouts PDFDocument53 pagesHandouts PDFSandeep ChallaNo ratings yet
- L26 27 PDFDocument35 pagesL26 27 PDFSandeep ChallaNo ratings yet
- Interpretation:: Lewis Base Lewis AcidDocument4 pagesInterpretation:: Lewis Base Lewis AcidSandeep ChallaNo ratings yet
- Interpretation:: CH NH BFDocument2 pagesInterpretation:: CH NH BFSandeep ChallaNo ratings yet
- 3 2PPDocument3 pages3 2PPSandeep ChallaNo ratings yet
- COMMWIP Assignment - Hemant VermaDocument3 pagesCOMMWIP Assignment - Hemant VermaSandeep ChallaNo ratings yet
- Assignment 2 3Document3 pagesAssignment 2 3Sandeep Challa0% (1)
- 3 2PPDocument3 pages3 2PPSandeep ChallaNo ratings yet
- B.tech Chemical DatabaseDocument5 pagesB.tech Chemical DatabaseSandeep ChallaNo ratings yet
- Distillation Column DesignDocument20 pagesDistillation Column DesignSandeep Challa100% (1)
- OrientDocument20 pagesOrientSandeep ChallaNo ratings yet
- DHDS ProcessDocument9 pagesDHDS ProcessSandeep ChallaNo ratings yet