You might also like
- Good Clinical Practice - Harshad MoreDocument24 pagesGood Clinical Practice - Harshad MoreHarshad MoreNo ratings yet
- Ich-Gcp & Schedule yDocument38 pagesIch-Gcp & Schedule ySahiti PendyalaNo ratings yet
- ICH E6-R3 GCP-Principles Draft 2021 0419Document7 pagesICH E6-R3 GCP-Principles Draft 2021 0419ramya sandraNo ratings yet
- Adverse Drug ReactionDocument15 pagesAdverse Drug ReactionPuji Arifianti RamadhanyNo ratings yet
- ELISADocument19 pagesELISAkeijiNo ratings yet
- Introduction To Clinical Research by SathDocument19 pagesIntroduction To Clinical Research by SathVignesh GaneshNo ratings yet
- ICH GCP & Indian Clinical Trial GuidelineDocument97 pagesICH GCP & Indian Clinical Trial GuidelineRanjeet PrasadNo ratings yet
- Farmakokinetika AdmeDocument42 pagesFarmakokinetika AdmeFerda Mahdalena100% (1)
- Blueprint For Clinical Research Sops FINALDocument41 pagesBlueprint For Clinical Research Sops FINALZeeshan Zafar0% (1)
- Ethical Issues in Clinical TrialsDocument16 pagesEthical Issues in Clinical TrialsPranjal KothaleNo ratings yet
- Ethics in Clinical TrialDocument28 pagesEthics in Clinical TrialRanjeet PrasadNo ratings yet
- Phases in Clinical TrialsDocument4 pagesPhases in Clinical TrialsManish SarasvatiNo ratings yet
- Monolisa HCV Ag-Ac UltraDocument4 pagesMonolisa HCV Ag-Ac UltraSantiagoAFNo ratings yet
- Research TechniquesDocument90 pagesResearch TechniquesTimothy Ian KooNo ratings yet
- Clinical Trials and Review & Approval of Clinical StudiesDocument55 pagesClinical Trials and Review & Approval of Clinical StudiesKinal MehtaNo ratings yet
- Immunodiagnostics: Dr. OyaroDocument32 pagesImmunodiagnostics: Dr. Oyaroodhiambo samwelNo ratings yet
- New Drugs & Clinical Trial RulesDocument32 pagesNew Drugs & Clinical Trial RulesSupriya Ghanekar PhadkeNo ratings yet
- Lecture 9 - POCT Blood GlucoseDocument43 pagesLecture 9 - POCT Blood GlucoseRama ArdianaNo ratings yet
- Lab Diagnosis of NeoplasiaDocument14 pagesLab Diagnosis of Neoplasiasapphiresia100% (6)
- Pharmacovigilance in Clinical Trials - PPTX FinalDocument20 pagesPharmacovigilance in Clinical Trials - PPTX Finalmina100% (1)
- GCP Flash CardDocument3 pagesGCP Flash CardSok YeeNo ratings yet
- Katherine Birch Specialist Testing of CSFDocument43 pagesKatherine Birch Specialist Testing of CSFmonday125No ratings yet
- Principles, Applications and InterpretationsDocument64 pagesPrinciples, Applications and InterpretationsAbbi Yanto ArtNo ratings yet
- Old Drugs For A New Use (Prescription)Document17 pagesOld Drugs For A New Use (Prescription)Karol Denisse Hernández BastidaNo ratings yet
- Skin and Soft Tissue InfectionsDocument8 pagesSkin and Soft Tissue InfectionsciaranNo ratings yet
- 4.10.17final Clinical Trials Talk.4.10.2017 - 300526 - 284 - 30426 - v1Document67 pages4.10.17final Clinical Trials Talk.4.10.2017 - 300526 - 284 - 30426 - v1Mohammed HammedNo ratings yet
- Sepsis Diagnosis and Monitoring: Fast Early ReliableDocument24 pagesSepsis Diagnosis and Monitoring: Fast Early ReliableI Nyoman Gede Semarajana100% (1)
- Introduction To Clinical ResearchDocument19 pagesIntroduction To Clinical ResearchVignesh GaneshNo ratings yet
- Auto Analyzer: Medical ElectronicsDocument12 pagesAuto Analyzer: Medical ElectronicsDd0% (1)
- Antifungal Drugs Powerpoint (M)Document70 pagesAntifungal Drugs Powerpoint (M)nk999999No ratings yet
- Serum Protein ElectrophoresisDocument9 pagesSerum Protein Electrophoresiskiedd_04100% (4)
- Biological AssaysDocument19 pagesBiological AssaysSubhash DhungelNo ratings yet
- 3 History and Principles of Good Clinical Practice 1Document24 pages3 History and Principles of Good Clinical Practice 1Larissa Miguel Severa100% (2)
- Nanoparticles in Cancer Therapy and DiagnosisDocument55 pagesNanoparticles in Cancer Therapy and Diagnosismimshin0% (1)
- Medical Law EthicsDocument30 pagesMedical Law EthicsImad AnwarNo ratings yet
- Aminoglycoside Dosing GuidelinesDocument3 pagesAminoglycoside Dosing GuidelinesaeromintNo ratings yet
- Clinical TrialDocument25 pagesClinical TrialHernawati Bagenda100% (1)
- Case Study Analysis (1,2,3,4) ENDOCRINOLOGYDocument3 pagesCase Study Analysis (1,2,3,4) ENDOCRINOLOGYDayledaniel Sorveto100% (1)
- Anameia PPT by Sandeep - FullDocument32 pagesAnameia PPT by Sandeep - Fullsravanthi100% (1)
- Allaergic Conjunctivitis PathophysiologyDocument25 pagesAllaergic Conjunctivitis PathophysiologyRabiu Hassan MusaNo ratings yet
- Scientist Viva AnswerDocument2 pagesScientist Viva Answermonday125No ratings yet
- Indian in Vitro Diagnostics Market Analysis 2018Document4 pagesIndian in Vitro Diagnostics Market Analysis 2018Neeraj ChawlaNo ratings yet
- Lecture 2 Introductiion 2022Document23 pagesLecture 2 Introductiion 2022Ahmed HamarnehNo ratings yet
- 2.5 Antibody ScreeningDocument5 pages2.5 Antibody ScreeningBALAJINo ratings yet
- ImmunoassayDocument7 pagesImmunoassaySanthosh PNo ratings yet
- Systemic Fungal InfectionDocument43 pagesSystemic Fungal InfectionDarian DavinNo ratings yet
- Critical Appraisal: DR A C J Hutchesson Chair of Examiners' Panel, Frcpath (Clinical Biochemistry)Document10 pagesCritical Appraisal: DR A C J Hutchesson Chair of Examiners' Panel, Frcpath (Clinical Biochemistry)monday125No ratings yet
- DR Cole Storage Disorders Hand OutDocument3 pagesDR Cole Storage Disorders Hand Outmonday125No ratings yet
- Phases of Clinical TrialDocument10 pagesPhases of Clinical TrialUmardin SaifyNo ratings yet
- EsrDocument5 pagesEsraftab ahmadNo ratings yet
- Genetic Polymorphism FixDocument63 pagesGenetic Polymorphism FixBiean gantengNo ratings yet
- Therapeutic Regimens in HIVDocument36 pagesTherapeutic Regimens in HIVGail HoadNo ratings yet
- Clinical Research ClassDocument110 pagesClinical Research ClassEreshNo ratings yet
- Subject Name: Pharmacology II, Subject Code: BP 503T Unit II Chapter-4: Plasma Volume Expanders by Dr. Varsha RajDocument21 pagesSubject Name: Pharmacology II, Subject Code: BP 503T Unit II Chapter-4: Plasma Volume Expanders by Dr. Varsha RajUbais AlamNo ratings yet
- Pulmonary DeliveryDocument25 pagesPulmonary DeliveryAhmed Osama ShalashNo ratings yet
- In Vitro MarketDocument59 pagesIn Vitro MarketmastbittuNo ratings yet
- National ART Clinical Guideline 2023 - 04 - 28 SignedDocument44 pagesNational ART Clinical Guideline 2023 - 04 - 28 SignedZimkita Zintle MakeleniNo ratings yet
- Introduction To TDMDocument48 pagesIntroduction To TDMKumara Swamy MNo ratings yet
- Investigators Responsibilities With GCPDocument16 pagesInvestigators Responsibilities With GCPLlosa JuneNo ratings yet
- Part 1: Introduction Part 2: Responsibilities by Role Part 3: Summary of Key PointsDocument13 pagesPart 1: Introduction Part 2: Responsibilities by Role Part 3: Summary of Key PointsFaye BaliloNo ratings yet
- Training Log - Hayya Life Science - Self Training - HLCDocument2 pagesTraining Log - Hayya Life Science - Self Training - HLCHerry SinambelaNo ratings yet
- Question Paper DSSSB Phamacist April 2015Document29 pagesQuestion Paper DSSSB Phamacist April 2015Sahil KumarNo ratings yet
- Tugas Terapi Modalitas Keperawatan Chronic Kidney Disease (CKD)Document7 pagesTugas Terapi Modalitas Keperawatan Chronic Kidney Disease (CKD)Winda WidyaNo ratings yet
- Design of Clinical TrialsDocument19 pagesDesign of Clinical TrialsSonu SharmaNo ratings yet
- Bioavailabilitas & BioekivalensiDocument19 pagesBioavailabilitas & BioekivalensiCandriya LaelNo ratings yet
- Pharmacovigilance Related Questions - 1585456120320Document10 pagesPharmacovigilance Related Questions - 1585456120320Saravanan RamNo ratings yet
- CDM Interview QuestionDocument46 pagesCDM Interview QuestionBheem Yadav100% (3)
- Potential Interview QuestionsDocument0 pagesPotential Interview QuestionsRahul PuriNo ratings yet
- Assignment For L3: L3 Dynamics in Various Actions and FunctionsDocument2 pagesAssignment For L3: L3 Dynamics in Various Actions and FunctionsRumana AliNo ratings yet
- Pharmacovigilance: A ReviewDocument4 pagesPharmacovigilance: A ReviewMahewash PathanNo ratings yet
- Diploma in Pharmacy YearSession 2022-2023-MAY-FINAL REGULAR Term 2 Term ReportDocument2 pagesDiploma in Pharmacy YearSession 2022-2023-MAY-FINAL REGULAR Term 2 Term Report2506sanjaydwivediNo ratings yet
- Guidance To Industry and Reviewers - Exploratory IND Studies PDFDocument16 pagesGuidance To Industry and Reviewers - Exploratory IND Studies PDFcool BunnNo ratings yet
- Regulatory Affairs PDFDocument12 pagesRegulatory Affairs PDFdrgdsw50% (2)
- Govt Approved D PHARMA Institutions 2020Document5 pagesGovt Approved D PHARMA Institutions 2020Hahah hzhNo ratings yet
- COVID Vaccines VAERS Data by Lot Number: Preliminary AnalysisDocument20 pagesCOVID Vaccines VAERS Data by Lot Number: Preliminary AnalysisAlberico MedeirosNo ratings yet
- Angell M. The Ethics of Clinical ResearchDocument9 pagesAngell M. The Ethics of Clinical ResearchMaria RivasNo ratings yet
- 10 1089@acm 2018 0046Document24 pages10 1089@acm 2018 0046rezteevicNo ratings yet
- Kajian Hukum Peran "Apoteker" Dalam Saintifikasi JamuDocument6 pagesKajian Hukum Peran "Apoteker" Dalam Saintifikasi JamuPetrisyia ReyvhonniNo ratings yet
- Saldo 02 NovDocument2 pagesSaldo 02 NovLuciana ReyesNo ratings yet
- Dialogue Between Pharmacist and PatientDocument2 pagesDialogue Between Pharmacist and PatientAbid Ali Khan90% (10)
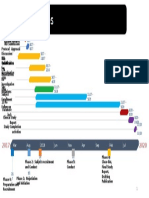
- Study Timeline ExampleDocument1 pageStudy Timeline ExamplePamela WolfeNo ratings yet
- BSPhar AY 2021-2022Document51 pagesBSPhar AY 2021-2022Crystal AnnNo ratings yet
- Jadad ScaleDocument4 pagesJadad ScaleLuis Henrique SalesNo ratings yet
- An Overview of The Common Technical Document (CTD) Regulatory DossierDocument5 pagesAn Overview of The Common Technical Document (CTD) Regulatory DossierNguyen Van TrangNo ratings yet
- Investigator HandbookDocument116 pagesInvestigator Handbookjack100% (1)
- Clinical Trial FinalDocument17 pagesClinical Trial Finalwondimnew WalleNo ratings yet
- Pharma CompanyDocument4 pagesPharma CompanyjennyNo ratings yet
- IPPCR 2023-2024 Syllabus FINAL 2-508Document2 pagesIPPCR 2023-2024 Syllabus FINAL 2-508asi basseyNo ratings yet
- Practical ScheduleDocument5 pagesPractical ScheduleJutt GeNo ratings yet
- Adverse Drug ReactionDocument10 pagesAdverse Drug ReactionNatasa KidisevicNo ratings yet