You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Class IX Science Why Do We Fall Ill - Lesson Plan PDocument16 pagesClass IX Science Why Do We Fall Ill - Lesson Plan PShailaja Mestry0% (2)
- Artculo FSC 2020 Estevez ColpasDocument44 pagesArtculo FSC 2020 Estevez ColpasFredy Colpas CastilloNo ratings yet
- Novel Composite Adsorbent For Adsorption of Urea: Linqi Shi, Yuzhong Zhang and Binglin HeDocument5 pagesNovel Composite Adsorbent For Adsorption of Urea: Linqi Shi, Yuzhong Zhang and Binglin HeFredy Colpas CastilloNo ratings yet
- Flotation of Sylvinite From Thakhek, Lao, P.D.R.: Chairoj Rattanakawin, Woraruethai Lakantha, and Ittirit KajaiDocument6 pagesFlotation of Sylvinite From Thakhek, Lao, P.D.R.: Chairoj Rattanakawin, Woraruethai Lakantha, and Ittirit KajaiFredy Colpas CastilloNo ratings yet
- Novel Composite Adsorbent For Adsorption of Urea: Linqi Shi, Yuzhong Zhang and Binglin HeDocument5 pagesNovel Composite Adsorbent For Adsorption of Urea: Linqi Shi, Yuzhong Zhang and Binglin HeFredy Colpas CastilloNo ratings yet
- Caballero EstevezDocument13 pagesCaballero EstevezFredy Colpas CastilloNo ratings yet
- Data in Brief PhenantrenompublicadoDocument6 pagesData in Brief PhenantrenompublicadoFredy Colpas CastilloNo ratings yet
- Ambiente y Agua Fredy ColpasDocument9 pagesAmbiente y Agua Fredy ColpasFredy Colpas CastilloNo ratings yet
- The State of The Cubic Equations of State: ReviewsDocument16 pagesThe State of The Cubic Equations of State: ReviewsFredy Colpas CastilloNo ratings yet
- Experimental Measurement and Correlation For Solubility o - 2013 - The Journal o PDFDocument6 pagesExperimental Measurement and Correlation For Solubility o - 2013 - The Journal o PDFFredy Colpas CastilloNo ratings yet
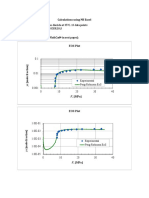
- System: Naphthalene + Carbon Dioxide At 35°C, 13 Data Points Experimental Data File: Nfcd35Sr.Da3 For Α = 4.451; Β = 5.79 Plots (Sample Calculation In Mathcad In Next Pages)Document4 pagesSystem: Naphthalene + Carbon Dioxide At 35°C, 13 Data Points Experimental Data File: Nfcd35Sr.Da3 For Α = 4.451; Β = 5.79 Plots (Sample Calculation In Mathcad In Next Pages)Fredy Colpas CastilloNo ratings yet
- The State of The Cubic Equations of State: ReviewsDocument16 pagesThe State of The Cubic Equations of State: ReviewsFredy Colpas CastilloNo ratings yet
- Holder Solubility of Anthracene and Phenanthrene Mixtures in Supercritical Carbon Dioxide.Document3 pagesHolder Solubility of Anthracene and Phenanthrene Mixtures in Supercritical Carbon Dioxide.Fredy Colpas CastilloNo ratings yet
- Estevez 2008 CES 63 4292 DBased Solub SCFCO2 Model Sparks PDFDocument10 pagesEstevez 2008 CES 63 4292 DBased Solub SCFCO2 Model Sparks PDFFredy Colpas CastilloNo ratings yet
- 2007 Tsang Activ Carbon PDFDocument15 pages2007 Tsang Activ Carbon PDFFredy Colpas CastilloNo ratings yet
- Dobrowolski 2010 PDFDocument8 pagesDobrowolski 2010 PDFFredy Colpas CastilloNo ratings yet
- Selvi 2001 PDFDocument3 pagesSelvi 2001 PDFFredy Colpas CastilloNo ratings yet
- Selvi 2001 PDFDocument3 pagesSelvi 2001 PDFFredy Colpas CastilloNo ratings yet
- Dobrowolski 2010 PDFDocument8 pagesDobrowolski 2010 PDFFredy Colpas CastilloNo ratings yet
- Paper InnovationDocument8 pagesPaper InnovationFredy Colpas CastilloNo ratings yet
- BituminosoDocument5 pagesBituminosoFredy Colpas CastilloNo ratings yet
- Jhon Lee Gas ReservesDocument26 pagesJhon Lee Gas ReservesMitsúMilagrosToroSayasNo ratings yet
- Phychem Lab Notes Ex 5Document3 pagesPhychem Lab Notes Ex 5Skye DiazNo ratings yet
- Modelado y Simulación Del Proceso de Desolventización-Tostado de Harina de Aceite de SojaDocument8 pagesModelado y Simulación Del Proceso de Desolventización-Tostado de Harina de Aceite de SojaROMMYNo ratings yet
- Guest AbbVie LLPS Dynochem Final 20210623Document28 pagesGuest AbbVie LLPS Dynochem Final 20210623Sasitharan MNo ratings yet
- 4 Ponchon Savarit MethodDocument47 pages4 Ponchon Savarit Methodivanlawms6745No ratings yet
- Oil - Program Vilter PDFDocument92 pagesOil - Program Vilter PDFRicardoNo ratings yet
- Brosur Witepsol PDFDocument25 pagesBrosur Witepsol PDFMuhammad Arnaj MadyanNo ratings yet
- Literature Review of Gas AbsorptionDocument5 pagesLiterature Review of Gas Absorptionafdtbbhtz100% (1)
- I. Title of Experiment: Equilibrium Phase Two Component II. Date of Experiment: Friday, 19 III. Purpose of ExperimentDocument7 pagesI. Title of Experiment: Equilibrium Phase Two Component II. Date of Experiment: Friday, 19 III. Purpose of ExperimentUtari Ika CahyaniNo ratings yet
- Biophysics Final QuestionsDocument4 pagesBiophysics Final Questionshadigy1001No ratings yet
- Chapter 2 - Wettability - Surface and Interfacial TensionDocument169 pagesChapter 2 - Wettability - Surface and Interfacial TensionNubasyer QallinsmanNo ratings yet
- Phase Rule: Dr.S.Ignatius Arockiam, Assistant Professor, Dept of ChemistryDocument24 pagesPhase Rule: Dr.S.Ignatius Arockiam, Assistant Professor, Dept of ChemistryMohamed IdrishNo ratings yet
- Absorption Lecture Note - DR Akinsiku PDFDocument7 pagesAbsorption Lecture Note - DR Akinsiku PDFGlory UsoroNo ratings yet
- FLUENT - Tutorial - VOF - Horizontal Film BoilingDocument16 pagesFLUENT - Tutorial - VOF - Horizontal Film BoilingBrilliand Tegar VerlambangNo ratings yet
- Reliable Level of Corrosion Inhibitor's Residual Concentration in Wet Gas-Condensate PipelinesDocument13 pagesReliable Level of Corrosion Inhibitor's Residual Concentration in Wet Gas-Condensate Pipelines陳冠宏No ratings yet
- CAP6 Whitson Phase BehaviorDocument21 pagesCAP6 Whitson Phase BehaviorMaría José MartínezNo ratings yet
- Hydrothermal Liquefaction Reactor DesignDocument90 pagesHydrothermal Liquefaction Reactor Designraymond tambunanNo ratings yet
- Journal of Materials Processing Technology: S.K. Choudhary, S. Ganguly, A. Sengupta, V. SharmaDocument10 pagesJournal of Materials Processing Technology: S.K. Choudhary, S. Ganguly, A. Sengupta, V. SharmaShivam SrivastavaNo ratings yet
- Exercise 33: This Is Your Last Free ExplanationDocument7 pagesExercise 33: This Is Your Last Free Explanation201903845 Astrid GutiérrezNo ratings yet
- Final Multiple Choice (Chemistry)Document13 pagesFinal Multiple Choice (Chemistry)wizett2No ratings yet
- Chemistry CH - 2 Part-IDocument4 pagesChemistry CH - 2 Part-IDr. Abdul Haq BalochNo ratings yet
- Spirax Sarco Steam Utilization DesignDocument66 pagesSpirax Sarco Steam Utilization DesignLee JianNo ratings yet
- PQT Chapter 9a Phase DiagramsDocument53 pagesPQT Chapter 9a Phase DiagramsDương Hữu PhươngNo ratings yet
- MEC451 - CH 03 - Part 3 - Closed System - ODL DiscussionDocument17 pagesMEC451 - CH 03 - Part 3 - Closed System - ODL DiscussionSalahuddin NorazmiNo ratings yet
- Kalina Cycle PDFDocument11 pagesKalina Cycle PDFcanscot50% (2)
- 6.23 Equation of State Prediction of Carbon Dioxide PropertiesDocument70 pages6.23 Equation of State Prediction of Carbon Dioxide Propertiessaulatlone100% (1)
- CH 09Document94 pagesCH 09Wong Li XuanNo ratings yet
- Dense Phase DepressurizationDocument2 pagesDense Phase DepressurizationSONWALYOGESHNo ratings yet
- 1 MolarMassVolatileLiquidDocument4 pages1 MolarMassVolatileLiquidkermithmmoguel50% (2)