Professional Documents
Culture Documents
Floege2016 PDF
Floege2016 PDF
Uploaded by
Carlos DominguezOriginal Title
Copyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
Floege2016 PDF
Floege2016 PDF
Uploaded by
Carlos DominguezCopyright:
Available Formats
Seminar
Primary glomerulonephritides
Jürgen Floege, Kerstin Amann
Lancet 2016; 387: 2036–48 Most glomerulonephritides, even the more common types, are rare diseases. They are nevertheless important since
Published Online they frequently affect young people, often cannot be cured, and can lead to chronic kidney disease, including end-
February 24, 2016 stage renal failure, with associated morbidity and cost. For example, in young adults, IgA nephropathy is the most
http://dx.doi.org/10.1016/
common cause of end-stage renal disease. In this Seminar, we summarise existing knowledge of clinical signs,
S0140-6736(16)00272-5
pathogenesis, prognosis, and treatment of glomerulonephritides, with a particular focus on data published between
Department of Nephrology
and Clinical Immunology, 2008 and 2015, and the most common European glomerulonephritis types, namely IgA nephropathy, membranous
University Hospital, Rheinisch glomerulonephritis, minimal change disease, focal segmental glomerulosclerosis, membranoproliferative
Westfälische Technische glomerulonephritis, and the rare complement-associated glomerulonephritides such as dense deposit disease and
Hochschule Aachen, Aachen,
C3 glomerulonephritis.
Germany (Prof J Floege MD);
and Department of
Nephropathology, Department Introduction Laboratory findings
of Pathology, University of Glomerulonephritides account for about 20% of chronic Clinically manifest glomerulonephritides exhibit
Erlangen-Nürnberg, Erlangen,
kidney disease cases in most countries and, unlike variable amounts of proteinuria or haematuria. Excretion
Germany (Prof K Amann MD)
major causes of chronic kidney disease such as diabetes of large amounts of albumin but no high-molecular-
Correspondence to:
Prof Jürgen Floege, Department and hypertension, they frequently affect young people weight proteins characterises minimal change disease
of Nephrology and Clinical and many carry a lifelong burden of chronic kidney in children, but this so-called selective proteinuria is
Immunology, Rheinisch disease. Indeed, in young adults, glomerulonephritides rarely present in adults. Increased urinary excretion of
Westfälische Technische
are the most frequent cause of end-stage renal disease. low-molecular-weight proteins such as alpha-1-
Hochschule University of Aachen,
D-52057 Aachen, Germany The glomerulonephritides are a heterogeneous group of microglobulin suggests that the primary glomerular
juergen.floege@rwth-aachen. diseases, most of which qualify as orphan diseases. injury has spread to the tubular system, impairing
de Consequently, few clinical trials exist. The 2012 the reabsorption of such proteins in the tubule,
guidelines by the Kidney Disease: Improving Global a prognostically adverse sign.2 In particular,
Outcomes (KDIGO) initiative on the treatment of oligosymptomatic glomerulonephritides such as IgA
glomerular diseases1 represent a major advance in nephropathy frequently exhibit some renal failure at
this specialty. first presentation. More specific laboratory tests, such as
the detection of antibodies to phospholipase A2 receptor
Clinical presentation (PLA2R), are strongly suggestive of a membranous
No specific clinical signs of glomerulonephritis exist, glomerulonephritis,3 whereas identification of cir-
which emphasises the need to keep this potential culating immune complexes, total IgA in serum, or anti-
diagnosis in mind. The clinical course of glomerulo- streptolysin O titres is no longer regarded as
nephritis is variable, ranging from chance findings in diagnostically helpful in most cases.
asymptomatic patients (eg, hypertension, proteinuria by
dipstick, haematuria, and raised serum creatinine
concentrations), to massive weight gain and oedema with
Search strategy and selection criteria
nephrotic syndrome, to rapidly progressive glomerulo-
nephritis with uraemia. Some clinical presentations can We searched PubMed, Embase, and the Cochrane Library
be suggestive of a particular diagnosis; in children and between October, 2008, and Oct 31, 2015, for the following
young adults, nephrotic syndrome suggests minimal search terms: “IgA-nephropathy”, “IgA glomerulonephritis”,
change disease or focal segmental glomerulosclerosis, “IgA nephritis”, “membranous nephropathy”, “membranous
whereas in older adults it suggests a membranous glomerulonephritis”, “minimal change disease”, “minimal
glomerulonephritis. Young oligosymptomatic adults with change nephrotic syndrome”, “focal segmental
hypertension or mild oedema might have IgA nephro- glomerulosclerosis”, “FSGS”, “membranoproliferative
pathy. In these cases, the onset of macrohaematuria glomerulonephritis”, “mesangiocapillary glomerulonephritis”,
within 1–2 days after a respiratory tract infection is “dense deposit disease”, “C3 nephropathy”,
suggestive of IgA nephropathy. Additional glomerulo- “C3 glomerulonephritis”, “anti-GBM disease”, “anti-GBM
nephritis signs and symptoms might arise according to nephritis”, “C1q nephropathy”, and “fibrillary
the chronic kidney disease stage, such as renal anaemia glomerulonephritis”. No language restriction was applied.
with fatigue or nausea and vomiting caused by uraemic From about 11 000 articles, we selected original research and
gastroenteritis. The classic signs of acute glomerulo- meta-analyses that provided evidence-based information
nephritis, such as oliguria, oedema, haematuria, about the cause, management, and treatment of various
headache, and flank pain that develop about 2 weeks after types of glomerulonephritis in human beings. Particular focus
a streptococcal pharyngitis, have become rare in most was placed on randomised controlled trials.
developed countries.
2036 www.thelancet.com Vol 387 May 14, 2016
Seminar
25
20
15
Patients (%)
10
0
ep e
ep or
di ge
sc ve
e
e
y
is
rs
is
is
s
ep us
di ial
s
sc d
ep d
iti
iti
on tiv
iti
iti
as
as
th
th
iti
os
os
os
lo te
on te
he
ro si
an
on e
on o
it
hr
hr
hr
hr
se
se
hr
pa
pa
ul as
ul ra
ul an
ru en
ph ten
ul ia
id
ler
ler
rst
Ot
ch
ep
e
er se
er oc
ylo
ro
ro
er br
m gm
te
ne er
m roli
um
m di
sn
m ass
ph
ph
m m
in
Am
p
glo l se
Hy
glo Me
pu
ne
ne
glo op
lo
glo osi
glo CA-
im
r
e
bu
ca
Lu
an
A
ic
in
C3 dep
AN
Fo
Ig
Tu
et
M
br
ab
em
e
ns
Di
M
De
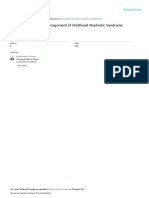
Glomerulonephritides
Figure 1: Kidney biopsy diagnoses in 2243 adult patients undergoing native kidney biopsy at the Division of Nephrology, Aachen University Hospital,
Aachen, Germany, between 1990 and 2013
ANCA=anti-neutrophil cytoplasmic antibodies. Data from Schlieper and colleagues (Schlieper G, Aachen University Hospital, Aachen, Germany, personal communication).
Differential diagnosis shown in the appendix. A renal biopsy is deemed a safe
Although nephrotic syndrome and rapidly progressive procedure if conditions are optimised (ie, experienced
renal failure are suggestive of a glomerulonephritis, physician, normal coagulation, no antiplatelet drugs for
most other clinical and laboratory findings carry a broad 7 days, normal blood pressure, no urinary tract infection,
range of differential diagnoses, including many non- and biopsy under ultrasound view with a 16–18 gauge
renal disorders (eg, cardiac oedema or lower urogenital needle).5 Perirenal haematoma is detected in 50–80% of
sources of haematuria). Phase-contrast microscopy of a patients and arteriovenous fistulas in up to 15% of
freshly voided urine specimen can help detect patients.6,7 These are usually asymptomatic and do not
dysmorphic red blood cells, in particular acanthocytes need intervention. Macroscopic haematuria occurs in up
(ie, red blood cells with one or more blebs of different to 8% of cases, but rarely results in bladder obstruction.6,7
size and shape protruding from a ring-shaped body),4 or Bleeding that necessitates transfusion, surgical
urinary red blood cell casts, both of which suggest a intervention, or nephrectomy occurs in less than 0·5% of
glomerular bleeding source. cases. In many centres, an overnight hospital stay is a
Establishment of whether a glomerulonephritis is requirement after a kidney biopsy, particularly if moderate-
primary or secondary is key in the work-up of a suspected to-advanced chronic kidney disease is present, because an
glomerulonephritis, because secondary glomerulo- assessment period of 8 h or less will miss about a third of
nephritis often necessitates treatment of the underlying clinically relevant complications.6
disorder rather than treatment for the glomerulonephritis.
Adequate diagnostic assessment should be considered Epidemiology
depending on the clinical situation (appendix). Figure 1 shows a typical range of diagnoses in a European See Online for appendix
renal biopsy cohort. Annual incidence has been estimated
Diagnostic investigations as 2·5 cases per 100 000 adults for IgA nephropathy,
A definitive diagnosis of glomerulonephritis requires a 1·2 per 100 000 for membranous glomerulonephritis,
kidney biopsy, which should be read by a nephro- 0·6–0·8 per 100 000 for minimal change disease and focal
pathologist. The biopsy core is assessed by light microscopy segmental glomerulosclerosis, and 0·2 per 100 000 for
and immunohistology, and often also by electron membranoproliferative glomerulonephritis.8 However,
microscopy. The biopsy will also provide important these percentages are usually underestimates because
information about acute inflammatory versus chronic they do not take into account people with asymptomatic
scarring changes. Common renal biopsy indications are glomerulonephritis variants, those whose glomerulo-
www.thelancet.com Vol 387 May 14, 2016 2037
Seminar
nephritis remits spontaneously, or, vice versa, those who
have advanced chronic kidney disease at first presentation Panel: Supportive measures in patients with
for whom a renal biopsy is no longer warranted. glomerulonephritis who are at risk of progressive loss of
Important regional differences in glomerulonephritis renal function
distribution exist; the percentage of IgA nephropathy Aim to implement all level A recommendations and as many
diagnoses is higher in Asian cohorts, whereas in cohorts level B recommendations as possible.
from the USA and Canada, focal segmental glomerulo-
sclerosis is more prevalent. Additionally, within one Level A recommendations
region, the distribution of glomerulonephritis types • Target seated systolic blood pressure 120–129 mm Hg
is associated with socioeconomic factors, with • Start ACE inhibitor or ARB treatment and up-titrate
wealthier countries exhibiting more IgA nephropathy dosage to reach the target systolic blood pressure and
and less wealthy countries having more cases of reduce proteinuria to less than 1 g/day
membranoproliferative glomerulonephritis.9 • Avoid dihydropyridine calcium-channel blockers unless
needed for blood pressure control (and then only after
Risk factors starting an ACE inhibitor or ARB)
In most glomerulonephritides, only a subgroup of • Control protein intake to about 0·8 g/kg per day
patients experiences progressive glomerular filtration Level B recommendations
rate (GFR) loss; such high-risk patients should be • Restrict sodium chloride intake or start diuretic treatment,
followed up by a nephrologist. These patients are or both
usually those with arterial hypertension, clinically • Control fluid intake
significant proteinuria (ie, >1 g/day in those with • Non-dihydropyridine calcium-channel blocker
IgA nephropathy or >3·5 g/day in those with treatment
focal segmental glomerulosclerosis, membranous • Control each component of the metabolic syndrome
glomerulonephritis, or membranoproliferative glom- • Aldosterone antagonist treatment (adapt dose to chronic
erulonephritis), and reduced estimated GFR at kidney disease stage)
glomerulonephritis diagnosis or histological presence • β-blocker treatment
of scars (ie, glomerulosclerosis or tubulointerstitial • Smoking cessation
fibrosis). In particular, proteinuria maintained over • Allopurinol treatment (controversial)
6–24 months is a potent predictor of outcome in the • Empiric sodium bicarbonate treatment, independent of
common glomerulonephritis types.10 Additional risk whether or not metabolic acidosis is present
factors for a progressive course include smoking11,12 and (controversial)
obesity,13 probably via increasing hypertension or
glomerular hyperfiltration, or both. Genetic factors Other measures to delay disease progression
such as the apolipoprotein L1 gene in African- • Avoid non-steroidal anti-inflammatory drugs if possible
Americans can also contribute to progression of (if not, use a maximum of once or twice weekly)
glomerulonephritides.14–16 Finally, coincident disorders • Avoid prolonged severe hypokalaemia
that damage the kidneys (eg, primary hypertension and • Correct vitamin D deficiency
diabetes mellitus) can also affect disease progression. • Control hyperphosphataemia and hyperparathyroidism
At the opposite end of the spectrum are those patients Modified from Wilmer and colleagues.17 ACE=angiotensin converting enzyme.
who only have mild renal failure, or renal failure that is ARB=angiotensin receptor blocker.
adequate for their age (most individuals will lose about
1 mL/min GFR per year after age 40 years), normo-
tension, minor proteinuria (below 1 g/day), or isolated In patients with nephrotic syndrome, the afore-
microhaematuria. These patients should be followed up mentioned measures should also include thrombosis
only periodically. prophylaxis. The concentration of serum albumin is
usually used as a surrogate parameter to assess the risk
Supportive therapy: one approach for all of thromboembolism. In particular, patients with
patients at risk of progressive disease membranous glomerulonephritis, especially those with
Supportive measures are not specific for patients with serum albumin below 20 g/L, are at an increased risk of
glomerulonephritis, but apply to all patients with thromboembolic events; the hazard ratio for thrombo-
proteinuric glomerular diseases who are at risk of embolism was 10·8 for patients with membranous
progressive disease (panel).17 Key measures include glomerulonephritis (95% CI 2·4–49·4) and 5·9 for those
antihypertensive, antiproteinuric, and dietary approaches with focal segmental glomerulosclerosis (1·3–27·9)
aimed at slowing down non-specific mechanisms that compared with those with IgA nephropathy.19 Risk:benefit
contribute to progression of renal disease. Such a instruments have been developed to establish who
For GN Tools see http:/www. comprehensive approach can slow progression of should receive anticoagulation; for example, GN Tools
gntools.com proteinuric glomerulonephritis substantially.18 for membranous glomerulonephritis.20
2038 www.thelancet.com Vol 387 May 14, 2016
Seminar
IgA nephropathy
A B
IgA nephropathy (figure 2) is the most common
glomerulonephritis in developed countries. It is usually an
oligosymptomatic glomerulonephritis, often discovered
coincidentally. Spontaneous remissions can occur.21 It is
regarded as an immune-mediated disease with deposition
of under-galactosylated, dimeric, or polymeric IgA in the
*
glomerular mesangium followed by the onset of
mesangioproliferative glomerulonephritis.22 IgG or IgA 100 µm
autoantibodies to the under-galactosylated IgA might also
contribute to IgA nephropathy,23,24 and both the circulating
C
concentrations of under-galactosylated IgA and of
autoantibodies are associated with IgA nephropathy
progression.24,25 Findings from a genome-wide association
study26 showed a pronounced genetic predisposition to IgA
nephropathy, with high prevalence in southeast Asia,
intermediate prevalence in the USA and Europe, and low
prevalence in Africa. Gene polymorphisms associated with
IgA nephropathy include complement factors and related 100 µm 100 µm
genes,27 HLA loci, and loci related to mucosal barrier
function and innate immunity.28,29 Figure 2: IgA nephropathy
(A) In IgA nephropathy, segmental areas (arrows) of mesangial hypercellularity and matrix expansion occur,
Major diagnostic advances characteristic of mesangioproliferative glomerulonephritis. Part of the glomerular tuft adheres to Bowman’s
capsule (white dashed oval), constituting the starting point of a secondary focal segmental glomerulosclerosis
Since many patients have a benign disease course or lesion. Tubulointerstitial damage with leucocyte infiltrates, tubular atrophy and fibrosis (arrowhead), and tubular
remit spontaneously,21 identification of those who will protein casts (asterix) is also present. PAS stain. (B) Other glomeruli in the same patient exhibit few pathological
progress to renal failure is crucial. In a French cohort,30 a abnormalities on light microscopy (PAS stain), but the characteristic mesangial granular IgA deposition (C) can be
three-point score based on proteinuria above 1 g/day, found in these glomeruli as well.
presence of hypertension, and histological changes in
the biopsy sample sensitively predicted the risk of death Therapeutic advances
or dialysis after 20 years (ranging from 4% in those with Uncertainty exists regarding immunosuppression in
a score of 0 to 64% with a score of 3). patients with IgA nephropathy who are at risk of
The Oxford-MEST classification of IgA nephropathy progressive disease.35 There is a consensus no longer to
comprises four histological parameters associated with a offer immunosuppression to patients with a GFR below
progressive course: glomerular mesangial hyper- 30 mL/min at presentation unless they also have a rapidly
cellularity; endocapillary hypercellularity; glomerular progressive glomerulonephritis course (see earlier).1
sclerosis or tuft adhesions; and tubular atrophy and Findings from a recent retrospective study36 suggest that
interstitial renal fibrosis affecting more than 25% of the corticosteroids might be beneficial for patients with a
section.31,32 Glomerular crescents were not included in GFR below 50 mL/min, particularly if proteinuria exceeds
this classification, possibly because patients who had a 3 g/day. In two randomised controlled trials (RCTs),
rapid decline to end-stage kidney disease were not angiotensin-converting enzyme (ACE) inhibition was
included in the original study. Additionally, in IgA compared with ACE inhibition plus high-dose oral
nephropathy, crescents can even occur in patients who prednisone in patients with IgA nephropathy who were at
are judged to have benign disease, such as isolated risk of progression (ie, proteinuria >1 g/day), but who had
microhaematuria.33 Such single crescents need to be a baseline GFR above 50 mL/min.37,38 Findings from both
differentiated from the rare clinical situation of crescentic studies showed that the combination was more effective
IgA nephropathy with glomerular necroses, crescents than ACE inhibition alone at preserving GFR. However,
affecting more than 50% of the glomeruli, and a clinical in both studies, patients were required to stop any pre-
course of rapidly progressive glomerulonephritis. In a existing renin-angiotensin system (RAS) blockers at least
Chinese study,34 70% of such patients developed end-stage 4 weeks before entering the trial, raising the concern that
renal disease after 5 years despite immunosuppressive some patients who entered the trial were already well
treatment. A serum creatinine concentration higher than controlled and in a low-risk proteinuria range (ie, below
600 μmol/L on admission defined a threshold beyond 1 g/day) with an RAS blocker alone. In a meta-analysis of
which immunosuppression might no longer be the role of corticosteroids in IgA nephropathy, findings
indicated. Rapidly progressive glomerulonephritis in IgA from all studies showed reductions in proteinuria, but
nephropathy must be distinguished from acute kidney several detected no benefits for GFR.39 In particular, the
injury induced by macrohaematuria with obstruction question of whether corticosteroids still exert a benefit if
and damage of tubules from glomerular bleeding. added after optimisation of supportive measures,
www.thelancet.com Vol 387 May 14, 2016 2039
Seminar
mycophenolate mofetil.46 Findings from an Italian
A B
multicentre RCT showed that the combination of
high-dose corticosteroids with azathioprine for 6 months
was not superior to steroids alone and again only
increased the frequency of adverse effects.47
The finding of intestinal hypersensitivity to many food
antigens in IgA nephropathy led to the suggestion of
enteric corticosteroids as a new treatment for IgA
nephropathy.48 In a pilot trial49 of budesonide given in a
formulation with preferential release in the terminal
ileum, albuminuria was reduced in IgA nephropathy; this
approach is being tested more formally in the multicentre
100 µm 100 µm
Nefigan phase 2 RCT (NCT01738035), and findings from
this trial are expected to be published soon. The value of a
C D tonsillectomy in patients with IgA nephropathy seems
questionable,50 and it is not routinely recommended.1 Fish
oil treatment is recommended by some, but not all,
physicians, if the patient can tolerate the taste.1
Controversies, uncertainties, and remaining research
questions
In view of increasing data suggesting that immuno-
suppression is not particularly effective in IgA nephro-
pathy, better understanding of the pathophysiology of the
disease, in particular the mucosa–kidney axis, is of
100 µm 100 µm imminent importance.51 Perhaps IgA nephropathy
exhibits more similarities to allergic than to autoimmune
Figure 3: Membranous glomerulonephritis diseases; the absence of suitable animal models hampers
The PAS stain (A) shows slightly thickened glomerular basement membranes and prominent podocytes. On
testing this theory.22 Clarification of several other aspects,
immunohistology, granular deposits of (B) IgG and (C) C3c can be found along the glomerular basement membrane,
and pronounced de-novo expression of (D) PLA2R is present on the podocytes. PLA2R=phospholipase A2 receptor. ranging from the role of complement in IgA nephropathy
to inhibition of growth factors for glomerular cells such
including intense RAS blockade, remains unresolved. as platelet-derived growth factor, might also lead to new
The sequence of first optimising supportive measures for therapeutic approaches for IgA nephropathy.22
3–6 months before considering corticosteroids in patients
with persistent proteinuria above 1 g/day and GFR greater Membranous glomerulonephritis
than 50 mL/min has been suggested by the guidelines.1 Membranous glomerulonephritis (figure 3) primarily
We tested this sequence in the STOP-IgAN RCT,40 in affects middle-aged and old adults and can occur secondary
which only patients who maintained proteinuria above to infections (eg, hepatitis B and C), tumours (see later52),
0·75 g/day after a comprehensive optimisation of systemic immune diseases (lupus erythematosus), or
supportive treatment were randomly assigned to receive some drugs (eg, gold and penicillamine). The disease
additional immunosuppression or to continue on often manifests as nephrotic syndrome with or without
supportive care only. Although we too noted a transient loss of GFR. The risk of thromboembolism in patients
reduction of proteinuria with immunosuppression, we with nephrotic syndrome is high (see earlier).
did not identify a benefit in the GFR endpoint at 3 years Findings from a landmark study in 20093 showed that
and adverse effects increased with immunosuppression 70–80% of primary membranous glomerulonephritis
compared with no immunosuppression.41 cases in white people represent autoimmune responses
Other immunosuppressive drugs, or combination against the M-type PLA2R, which can be detected on
immunosuppressive treatment, are not recommended in glomerular podocytes and in subepithelial immune
patients with high-risk IgA nephropathy except for those deposits. Subsequently, findings from a genome-wide
rare patients with a rapidly progressive glomerulo- association study showed that particular polymorphisms
nephritis course (see earlier).1 Even though benefits of of the PLA2R gene combined with HLA-DQA1 poly-
mycophenolate mofetil have been described in a small morphisms increased the risk for primary membranous
RCT in Chinese patients with IgA nephropathy,42 such glomerulonephritis by up to 79 times.53 Major pathogenic
benefits have not been noted in similarly small RCTs in epitopes of PLA2R have been identified54,55 and might
white people.43–45 Additionally, several cases of lethal facilitate the design of new therapeutic approaches. A less
pneumocystis pneumonia occurred in patients with IgA common autoantigen is thrombospondin type-1 domain-
nephropathy with reduced GFR who were receiving containing 7A (THSD7A).56 Other autoantigens are likely
2040 www.thelancet.com Vol 387 May 14, 2016
Seminar
to be discovered, establishing primary membranous
A B
glomerulonephritis as a family of autoimmune disorders.
Autoantigens or exogenous antigens in the rare situation
of neonatal membranous glomerulonephritis include
neutral endopeptidase and bovine serum albumin.57
Major diagnostic advances
Assays for circulating PLA2R antibodies rapidly change
the diagnostic approach to membranous glomerulo-
nephritis. Circulating PLA2R antibodies seem to be
specific for membranous glomerulonephritis;58 however,
they can also occur in secondary membranous
glomerulonephritis. Their titre is associated with disease 100 µm 4 µm
activity and predicts prognosis and response to
treatment,59–62 the disease course after treatment,63 and Figure 4: Minimal change disease
recurrences after kidney transplantation.64 A decrease in (A) By light microscopy (PAS stain), no substantial abnormalities of the glomerular structure are noted.
antibody titres during treatment often precedes (B) Electron microscopy shows widespread effacement of podocyte foot processes (arrows) in the absence of
electron-dense or fibrillary deposits.
proteinuria response65 and can thus help to differentiate
patients who respond to treatment from those who are Immunosuppressive treatment for membranous
refractory to treatment. The finding of PLA2R on glomerulonephritis has so far relied on alkylating agents
podocytes on renal biopsy can also aid in the diagnosis plus corticosteroids or calcineurin inhibitors. In British
of membranous glomerulonephritis. In particular, high-risk patients (ie, those with impaired GFR at
patients who are negative for PLA2R or THSD7A baseline), the combination of prednisolone and
autoantibodies exhibit a high probability of spontaneous chlorambucil delayed GFR loss significantly better than
remission but should be screened for malignancy.66 In a ciclosporin or supportive treatment, but at the cost of
2014 meta-analysis, the most common malignancies more adverse effects.70 By contrast, mycophenolate
associated with membranous glomerulonephritis were mofetil plus RAS blockers did not induce more partial
lung carcinomas (22 [26%] of 85 cases), followed by or full remissions in patients with membranous
prostate carcinomas (13 cases [15%]), haematological glomerulonephritis than RAS blockers alone.71 Findings
malignancies (12 cases [14%]), and colorectal tumours from a meta-analysis72 showed that alkylating agents
(nine cases [11%]).52 (mostly cyclophosphamide) plus corticosteroids protect
GFR, that ciclosporin and mycophenolate mofetil are
Major therapeutic advances not superior to this combination, and that long-term
Spontaneous remission occurs in up to a third of effects of tacrolimus or adrenocorticotropic hormone,
patients, after a mean of 14 months, even if the initial both of which also reduce proteinuria in membranous
presentation includes massive proteinuria.67 This finding glomerulonephritis, are not well established. A newer
supports the policy of waiting for 6 months and asse- therapeutic option is rituximab; about two-thirds
ssing the pattern in proteinuria before considering of patients with nephrotic membranous glom-
immunosuppression unless a rapid loss of GFR is erulonephritis achieve partial or full remission with
present or life-threatening complications of nephrotic rituximab.73 However, in a large retrospective study,
syndrome occur.1 In a Dutch RCT,68 this approach was rituximab was not superior to alkylating agents or
tested by randomly assigning 26 patients with nephrotic calcineurin inhibitors at inducing proteinuria remission
syndrome with a normal GFR to early immuno- at 12 months.74
suppression or waiting with immunosuppression until
serum creatinine concentration had increased by 25% or Controversies, uncertainties, and remaining research
more. The two treatment groups did not differ in questions
remission rates, the course of GFR loss, or complications. The search is on to clarify the pathogenesis of
The same Dutch group retrospectively analysed membranous glomerulonephritis in the 20–30% of
the outcome of 254 patients with membranous patients who are negative for PLA2R and THSD7A, as
glomerulonephritis.69 Immunosuppression was only well as the pathogenesis of membranous lupus nephritis.
given if GFR decreased or serious complications of the Animal models mimicking the human autoimmune
nephrotic syndrome occurred. After 10 years, seven situation are in development and should help in the
patients (3%) had developed end-stage kidney disease, design of new therapeutic approaches. Additionally, we
25 (10%) had died, 52 (20%; 95% CI 44–60) had achieved still do not have formal proof that therapeutic decisions
full remission, and 90 (35%; 85–94) a partial remission of can be made on the basis of autoantibody titres. RCTs
their nephrotic syndrome. These data support a watchful studying the effects of B-cell-targeted treatment are
waiting approach in most patients. ongoing (eg, MENTOR, NCT01180036).
www.thelancet.com Vol 387 May 14, 2016 2041
Seminar
A B C
100 µm 100 µm 100 µm
Figure 5: Focal segmental glomerulosclerosis
(A) Light microscopy (PAS stain) shows a segment of the glomerular tuft, which is collapsed and scarred resulting in adhesion of the glomerular tuft with Bowman‘s
capsule (arrow). (B and C) Parietal epithelial cell markers are sensitive markers for the detection of focal segmental glomerulosclerosis lesions. Serial sections of a
glomerulus with a lesion at the glomerular tip are shown. The PAS section (B) shows a glomerulus with minimal sclerosis. The glomerular tuft protrudes into the
tubular lumen (arrow), but no marked sclerosis or synechiae are visible in this section. Immunostaining (C) of the extracellular matrix produced by parietal epithelial
cells (red staining) and the parietal epithelial cell marker annexin A3 (green) clearly stain the lesion (arrow). (B and C) kindly provided by Bart Smeets, Nijmegen,
Netherlands, and Marcus Moeller, Aachen, Germany.
Minimal change disease and focal segmental hyperfiltration and hypertrophy; for example, in obesity
glomerulosclerosis or situations of reduced nephron mass (eg, premature
Minimal change disease (figure 4) is the most common birth or surgical removal of significant kidney mass).
disease underlying childhood nephrotic syndrome, but Renal risk variants in APOL1 contribute to focal
also manifests in adults. Secondary forms occur in segmental glomerulosclerosis progression in black
diseases such as atopy or malignancy, particularly in patients, regardless of diabetes status.14–16 Finally, focal
lymphomas. Minimal change disease is occasionally segmental glomerulosclerosis can also result from toxic
accompanied by glomerular IgA deposits, and this effects of drugs (eg, anabolic steroids)80 or viral infections
situation should not be confused with IgA nephropathy.75 (eg, HIV), inducing a collapsing variant of focal
Whether minimal change disease and focal segmental segmental glomerulosclerosis.
glomerulosclerosis represent different manifestations of
one disease (with minimal change disease potentially Major diagnostic advances
progressing to focal segmental glomerulosclerosis) or Glomerular permeability factors in minimal change
two different diseases is unknown. They are both covered disease include angiopoietin-like-4,81 and in focal
jointly here, since therapeutic studies often do not segmental glomerulosclerosis they might include
differentiate between them. soluble urokinase plasminogen activator receptor
Focal segmental glomerulosclerosis is a confusing (suPAR),82 cardiotrophin-like cytokine-1,83 and others.84
term because it is used by pathologists both to describe a Only suPAR has been assessed in several clinical studies.
glomerular scar (figure 5), which can result from almost Raised circulating suPAR concentrations specifically
any injury affecting the kidneys, and clinically to denote a identified patients with primary focal segmental
family of glomerular diseases. Central to the pathogenesis glomerulosclerosis in an initial study,82 but subsequent
is damage to podocytes, resulting in their loss. A second studies did not confirm this finding experimentally85 or
central event is activation of parietal epithelial cells on clinically.86–90 Rather, non-specific increases of suPAR
Bowman’s capsule, which then migrate onto the serum concentrations occur in patients with reduced
glomerular tuft to replace or displace podocytes.76 Once GFR or ongoing inflammation, or both. Thus, suPAR
on the glomerular tuft, parietal cells, unlike podocytes, measurements are not recommended in the differential
are unable to produce sufficient vascular endothelial diagnosis of nephrotic syndrome.
growth factor, and this triggers endothelial problems, Another potential diagnostic and therapeutic (see
with collapse and scarring of the affected capillary.77 The later) target is the co-stimulatory protein B7-1 (CD80).
finding of parietal cell activation markers can help to Normally macrophage derived, this molecule is found
distinguish focal segmental glomerulosclerosis from on podocytes in proteinuric diseases. Soluble urinary
minimal change disease (figure 5).78 CD80 increased in patients with nephrotic syndrome
Minimal change disease is assumed to result from with minimal change disease, but was low in patients
circulating permeability factors (see later).79 Within the in remission from minimal change disease and in
clinical focal segmental glomerulosclerosis family, one those with focal segmental glomerulosclerosis.91
group relates to circulating permeability-inducing factors In view of the growing list of podocyte molecules that
or mutations of podocyte proteins, both of which lead to can be mutated in focal segmental glomerulosclerosis, an
nephrotic syndrome mostly in children or young adults. important issue is whether genetic testing should be
Another group of focal segmental glomeruloscleroses done once a patient is diagnosed with primary focal
are secondary, adaptive forms, related to glomerular segmental glomerulosclerosis. Guidelines argue against
2042 www.thelancet.com Vol 387 May 14, 2016
Seminar
this unless a family history of focal segmental glomerulo- immunosuppressive drugs were discontinued shortly
sclerosis is present.1 This recommendation is supported after randomisation. At 1 year, 17 (71%) of 24 patients
by findings from a US study in non-familial focal who received rituximab had relapsed compared with
segmental glomerulosclerosis, in which mutations in 23 (96%) of 24 patients receiving placebo. Similar
commonly affected genes (NPHS2, TRPC6, ACTN4, findings were noted in children receiving a single dose
INF2, and PLCE1) were found in four of 28 children and of rituximab only108 and in adults.109–111 Side-effects were
none of 37 adults.92 By contrast, in families with steroid- generally mild to moderate, but occurred more
resistant nephrotic syndrome, a single-gene cause was frequently with rituximab than with placebo.107 One
detected in 526 (30%) of 1783 patients whose disease possible explanation for the efficacy of rituximab in
manifested before age 25 years.93 In adults, mutations of minimal change disease might be a direct effect of
the collagen IV alpha 3–5 genes, which also underlie rituximab on podocytes.112 Rituximab-resistant patients
Alport syndrome, have been identified in many patients with nephrotic syndrome responded to ofatumumab,
with focal segmental glomerulosclerosis.94 Additional another CD20 antibody.113
genetic causes are emerging.95–100 In a case series of five patients with primary or
recurrent focal segmental glomerulosclerosis after
Major therapeutic advances kidney transplantation,114 abatacept, an inhibitor of B7-1
Treatment of minimal change disease and focal segmental (see earlier), induced remissions of proteinuria, possibly
glomerulosclerosis relies on corticosteroids, with by reducing podocyte migration. In all cases, B7-1
prolonged high-dose treatment needed in focal segmental overexpression was identified in podocytes before
glomerulosclerosis.1 Corticosteroids, at least in focal treatment. However, others have not confirmed the
segmental glomerulosclerosis, might exert their effect by overexpression of B7-1115 or similar effects of abatacept,116
inducing expansion of myeloid-derived suppressor cells.101 and thus further confirmation should be awaited before
Treatment is difficult in steroid-resistant or frequently considering abatacept in patients with focal segmental
relapsing patients. In an RCT, ciclosporin was compared glomerulosclerosis. In another recent study,117 the value
with a combination of steroid pulses plus mycophenolate of galactose or adalimumab treatment in focal segmental
mofetil in 138 patients with steroid-resistant primary focal glomerulosclerosis was assessed, but findings were
segmental glomerulosclerosis.102 All endpoints, in inconclusive because of recruitment problems.
particular remission, did not differ between the two
approaches. However, even this large RCT is likely to have Controversies, uncertainties, and remaining research
been underpowered to detect therapeutic differences in questions
view of the heterogeneity of focal segmental glomerulo- One of the most important research issues in both
sclerosis pathogenesis and thus potential differences in minimal change disease and focal segmental glomerulo-
therapeutic responses.103 In another RCT, 6-monthly sclerosis is the identification of circulating permeability
cyclophosphamide boluses was compared with tacrolimus factors since this is likely to change our therapeutic
in adults with steroid-dependent minimal change approaches radically. Alternatively, a better understanding
disease.104 Both treatments induced complete remission of what mediates the damage to podocytes and activation
in over 75% of patients without major differences between of parietal glomerular epithelial cells in focal segmental
treatment groups. In an Indian RCT,105 ciclosporin was glomerulosclerosis is likely to result in new treatments. If
compared with tacrolimus (both combined with such activation is indeed a common pathway in various
corticosteroids) in paediatric steroid-resistant nephrotic causes of focal segmental glomerulosclerosis, it might be
syndrome; similar efficacy in inducing remission was particularly attractive for future RCTs.
noted, but relapses occurred 4·5 times more often with
ciclosporin and more cosmetic adverse effects occurred Membranoproliferative glomerulonephritides
with ciclosporin, an important consideration in children Membranoproliferative glomerulonephritis is a morpho-
and young adults. Finally, findings from a Cochrane logical pattern of injury and strictly speaking not a
review106 on immunosuppression in adults with focal disease. The term is likely to be phased out over time in
segmental glomerulosclerosis showed that ciclosporin favour of a more cause-specific nomenclature. Indeed,
combined with low-dose prednisolone can induce at least the existence of a primary, idiopathic membranopro-
partial remission of nephrotic syndrome, but that the liferative glomerulonephritis type I (mesangiocapillary
long-term benefits of preserving GFR are not well glomerulonephritis type I) has been questioned.118
established. The review advises against widespread use of Membranoproliferative glomerulonephritis type I has
alkylating agents in such patients. become a rare disease in most developed countries. About
Rituximab is increasingly used in minimal change a quarter of patients have an underlying hepatitis B or C
disease and focal segmental glomerulosclerosis. In an infection.119 Another quarter exhibit a monoclonal
RCT107 in children with frequently relapsing or steroid- gammopathy or lymphomas.119
dependent nephrotic syndrome, rituximab (four doses of Older electron-microscopy-based classification
375 mg/m²) was compared with placebo. All other systems also included membranoproliferative
www.thelancet.com Vol 387 May 14, 2016 2043
Seminar
glomerulonephritis type II (dense deposit disease) and Controversies, uncertainties, and remaining research
type III. A new terminology that distinguishes questions
immunoglobulin-mediated membranoproliferative The identification of specific causes underlying mem-
glomerulonephritis (ie, the former type I mentioned branoproliferative glomerulonephritis and C3 glomer-
earlier) from complement-mediated membranopro- ulopathies should hopefully allow rational treatment in
liferative glomerulonephritis has been proposed most cases. Studies of the rare so-called idiopathic
(appendix).120 The latter group, termed C3 membranoproliferative glomerulonephritis type I cases
glomerulopathy, is characterised by defects in the should be intensified to identify infectious or
alternative pathway of complement, in particular of other causes.
factor H,121 or autoantibodies to complement-regulatory
proteins (so-called C3 nephritic factors) leading to Rare glomerulonephritides
increased complement activation. The group includes Anti-glomerular basement membrane disease is a rare
dense deposit disease and C3 glomerulonephritis autoimmune glomerulonephritis affecting all age groups
(appendix).122–125 The two entities are distinguished on that develops as a result of conformational changes in
the basis of the immunohistological pattern of C3 and the α3NC1 and α5NC1 subunits of collagen IV in the
the electron microscopy detection of ribbon-like glomerular basement membrane followed by inter-
electron-dense deposits in the glomerular basement molecular and intra-molecular epitope spreading.135,136
membrane in dense deposit disease, versus deposits of This disease can occur as renal limited disease or with
usual density in C3 glomerulonephritis. Compared lung haemorrhage (Goodpasture disease). Symptoms
with patients with C3 glomerulonephritis, those with usually last only a few weeks, and progression within
dense deposit disease are usually younger and have days can also occur. Older patients usually exhibit milder
lower C3 serum concentrations, more extensive renal disease and, as in younger individuals, outcomes depend
damage, and a higher risk of GFR loss.126 High- on initial estimated GFR.137 Circulating antibodies to
throughput genetic analysis enabled identification of glomerular basement membrane collagen IV are
the defect underlying C3 glomerulonephritis in characteristic. However, 5% of patients are negative for
13 (43%) of 30 cases.127 Recurrence after kidney circulating antibodies but have linear IgG deposition
transplantation occurs in about 60–70% of patients along the glomerular basement membrane.138 Anti-
with C3 glomerulonephritis and more than 90% of glomerular basement membrane disease can occur
those with dense deposit disease.128 together with membranous glomerulonephritis, in
which case the renal manifestations tend to be milder
Major therapeutic advances than in pure anti-glomerular basement membrane
No new RCTs in membranoproliferative glomer- disease.139 Treatment of anti-glomerular basement
ulonephritis type I have been published in the past membrane disease should be started rapidly, using
5 years, and the KDIGO guidelines,1 which recommend corticosteroids, cyclophosphamide, and plasmapheresis.1
combined treatment with corticosteroids plus either Plasmapheresis seems to be essential, since
mycophenolate mofetil or cyclophosphamide in patients corticosteroids plus cyclophosphamide alone did not
with nephrotic syndrome with progressive GFR loss, are improve renal outcomes.140 Rituximab can help in
based on weak evidence. inducing disease remission.141 If patients present with
Individual patients with single dense deposit disease end-stage renal disease without pulmonary involvement,
or C3 glomerulonephritis were given eculizumab, a immunosuppression is no longer recommended.1
C5 antibody.129,130 Clinical symptoms and histological Although the disease can recur after kidney
findings improved in some patients. After 1 year of transplantation, survival of the allograft is not different
eculizumab, deposition of eculizumab was noted in from other diseases.142
glomeruli in a pattern resembling monoclonal IgG C1q nephropathy is a rare glomerulonephritis charac-
deposition disease.131 The relevance of this finding is terised by dominant or co-dominant mesangial or
unknown. Two patients with dense deposit disease with glomerular capillary C1q deposits. In a large case
autoantibodies against C3b or factor H received kidney series,143 clinical presentations ranged from minimal
transplants successfully without rapid disease recurrence urinary findings to nephrotic syndrome, and histological
under a special protocol of immunosuppression before findings from those suggestive of minimal change
and after transplantation.132 In another case, expression disease or focal segmental glomerulosclerosis to
of a hybrid protein (CFHR2-CFHR5) stabilised immune complex glomerulonephritis. In particular,
C3 convertase, and the resulting increased complement patients with pathological abnormalities consistent
activation could be stopped by soluble complement with focal segmental glomerulosclerosis progressed
receptor 1 only.133 Benefits from more non-specific to dialysis.
immunosuppression with mycophenolate mofetil plus Fibrillary glomerulonephritis is another rare disease of
corticosteroids have been described in patients with unknown cause associated with malignancies, para-
C3 glomerulonephritis.134 proteinaemias, and other autoimmune diseases.144
2044 www.thelancet.com Vol 387 May 14, 2016
Seminar
Proteinuria is often nephrotic. Half of patients progress 14 Parsa A, Kao WH, Xie D, et al, for the AASK Study Investigators
to end-stage renal disease, with frequent recurrence and the CRIC Study Investigators. APOL1 risk variants, race, and
progression of chronic kidney disease. N Engl J Med 2013;
after transplantation. Whether immunosuppression, in 369: 2183–96.
particular rituximab, affects the course of this disease 15 Kopp JB, Winkler CA, Zhao X, et al, for the FSGS-CT Study
is unknown.145 Consortium. Clinical features and histology of apolipoprotein
L1-associated nephropathy in the FSGS Clinical Trial.
J Am Soc Nephrol 2015; 26: 1443–48.
Conclusions 16 Freedman BI, Kopp JB, Langefeld CD, et al. The apolipoprotein L1
A former, descriptive glomerulonephritis classification, (APOL1) gene and nondiabetic nephropathy in African Americans.
J Am Soc Nephrol 2010; 21: 1422–26.
based largely on histological patterns, is increasingly 17 Wilmer WA, Rovin BH, Hebert CJ, Rao SV, Kumor K, Hebert LA.
being replaced on the basis of new pathogenic insights. Management of glomerular proteinuria: a commentary.
This change in classification has led to more and J Am Soc Nephrol 2003; 14: 3217–32.
more cause-driven treatments being developed. The 18 Ruggenenti P, Perticucci E, Cravedi P, et al. Role of remission
clinics in the longitudinal treatment of CKD. J Am Soc Nephrol
infrequency of most glomerulonephritides and the 2008; 19: 1213–24.
increasing knowledge about cause and treatment implies 19 Barbour SJ, Greenwald A, Djurdjev O, et al. Disease-specific risk of
that such patients should be followed up in specialised venous thromboembolic events is increased in idiopathic
glomerulonephritis. Kidney Int 2012; 81: 190–95.
centres, which, in turn, will facilitate RCTs, which are 20 Lee T, Biddle AK, Lionaki S, et al. Personalized prophylactic
desperately needed in nephrology.146 anticoagulation decision analysis in patients with membranous
nephropathy. Kidney Int 2014; 85: 1412–20.
Contributors
21 Gutierrez E, Zamora I, Ballarin JA, et al. Long-term outcomes of
JF and KA contributed to data collection, data analysis, data
IgA nephropathy presenting with minimal or no proteinuria.
interpretation, and writing of the manuscript. J Am Soc Nephrol 2012; 23: 1753–60.
Declaration of interests 22 Floege J. The pathogenesis of IgA nephropathy: what is new and
JF has received personal fees from Pharmalink, Amgen, Sanofi, how does it change therapeutic approaches? Am J Kidney Dis 2011;
Vifor/Fresenius, Chugai, and Bristol-Myers Squibb. KA declares no 58: 992–1004.
competing interests. 23 Suzuki H, Fan R, Zhang Z, et al. Aberrantly glycosylated IgA1 in
IgA nephropathy patients is recognized by IgG antibodies with
Acknowledgments restricted heterogeneity. J Clin Invest 2009; 119: 1668–77.
This work was supported by the German Research Foundation 24 Berthoux F, Suzuki H, Thibaudin L, et al. Autoantibodies targeting
(Deutsche Forschungsgemeinschaft) grant SFB TRR57, project p25. galactose-deficient IgA1 associate with progression of IgA
References nephropathy. J Am Soc Nephrol 2012; 23: 1579–87.
1 KDIGO. KDIGO clinical practice guideline for glomerulonephritis. 25 Zhao N, Hou P, Lv J, et al. The level of galactose-deficient IgA1 in
Kidney Int 2012; 2 (suppl 2): 139–274. the sera of patients with IgA nephropathy is associated with disease
2 van den Brand JA, Hofstra JM, Wetzels JF. Low-molecular-weight progression. Kidney Int 2012; 82: 790–96.
proteins as prognostic markers in idiopathic membranous 26 Kiryluk K, Li Y, Sanna-Cherchi S, et al. Geographic differences in
nephropathy. Clin J Am Soc Nephrol 2011; 6: 2846–53. genetic susceptibility to IgA nephropathy: GWAS replication study
3 Beck LH Jr, Bonegio RG, Lambeau G, et al. M-type phospholipase and geospatial risk analysis. PLoS Genet 2012; 8: e1002765.
A2 receptor as target antigen in idiopathic membranous 27 Zhu L, Zhai YL, Wang FM, et al. Variants in complement factor H
nephropathy. N Engl J Med 2009; 361: 11–21. and complement factor H-related protein genes, CFHR3 and
4 Kohler H, Wandel E, Brunck B. Acanthocyturia—a characteristic CFHR1, affect complement activation in IgA nephropathy.
marker for glomerular bleeding. Kidney Int 1991; 40: 115–20. J Am Soc Nephrol 2015; 26: 1195–204.
5 Nicholson ML, Wheatley TJ, Doughman TM, et al. A prospective 28 Li M, Foo JN, Wang JQ, et al. Identification of new susceptibility
randomized trial of three different sizes of core-cutting needle for loci for IgA nephropathy in Han Chinese. Nat Commun 2015;
renal transplant biopsy. Kidney Int 2000; 58: 390–95. 6: 7270.
6 Whittier WL, Korbet SM. Timing of complications in percutaneous 29 Kiryluk K, Li Y, Scolari F, et al. Discovery of new risk loci for IgA
renal biopsy. J Am Soc Nephrol 2004; 15: 142–47. nephropathy implicates genes involved in immunity against
intestinal pathogens. Nat Genet 2014; 46: 1187–96.
7 Hergesell O, Felten H, Andrassy K, Kühn K, Ritz E. Safety of
ultrasound-guided percutaneous renal biopsy-retrospective analysis 30 Berthoux F, Mohey H, Laurent B, Mariat C, Afiani A, Thibaudin L.
of 1090 consecutive cases. Nephrol Dial Transplant 1998; 13: 975–77. Predicting the risk for dialysis or death in IgA nephropathy.
J Am Soc Nephrol 2011; 22: 752–61.
8 McGrogan A, Franssen CF, de Vries CS. The incidence of primary
glomerulonephritis worldwide: a systematic review of the literature. 31 Cattran DC, Coppo R, Cook HT, et al. The Oxford classification of
Nephrol Dial Transplant 2011; 26: 414–30. IgA nephropathy: rationale, clinicopathological correlations, and
classification. Kidney Int 2009; 76: 534–45.
9 Johnson RJ, Hurtado A, Merszei J, Rodriguez-Iturbe B, Feng L.
Hypothesis: dysregulation of immunologic balance resulting from 32 Coppo R, Troyanov S, Bellur S, et al, for the VALIGA study of the
hygiene and socioeconomic factors may influence the epidemiology ERA-EDTA Immunonephrology Working Group. Validation of the
and cause of glomerulonephritis worldwide. Am J Kidney Dis 2003; Oxford classification of IgA nephropathy in cohorts with different
42: 575–81. presentations and treatments. Kidney Int 2014; 86: 828–36.
10 Barbour SJ, Cattran DC, Espino-Hernandez G, Hladunewich MA, 33 Liu H, Peng Y, Liu Y, et al. Renal biopsy findings of patients
Reich HN. Identifying the ideal metric of proteinuria as a predictor presenting with isolated hematuria: disease associations.
of renal outcome in idiopathic glomerulonephritis. Kidney Int 2015; Am J Nephrol 2012; 36: 377–85.
88: 1392–401. 34 Lv J, Yang Y, Zhang H, et al. Prediction of outcomes in crescentic
11 Orth SR, Ritz E, Schrier RW. The renal risks of smoking. Kidney Int IgA nephropathy in a multicenter cohort study. J Am Soc Nephrol
1997; 51: 1669–77. 2013; 24: 2118–25.
12 Yamaguchi M, Ando M, Yamamoto R, et al. Smoking is a risk factor 35 Floege J, Feehally J. Treatment of IgA nephropathy and
for the progression of idiopathic membranous nephropathy. Henoch-Schonlein nephritis. Nat Rev Nephrol 2013; 9: 320–27.
PLoS One 2014; 9: e100835. 36 Tesar V, Troyanov S, Bellur S, et al, for the VALIGA study of the
13 Bonnet F, Deprele C, Sassolas A, et al. Excessive body weight as a new ERA-EDTA Immunonephrology Working Group. Corticosteroids in
independent risk factor for clinical and pathological progression in IgA nephropathy: a retrospective analysis from the VALIGA study.
primary IgA nephritis. Am J Kidney Dis 2001; 37: 720–27. J Am Soc Nephrol 2015; 26: 2248–58.
www.thelancet.com Vol 387 May 14, 2016 2045
Seminar
37 Lv J, Zhang H, Chen Y, et al. Combination therapy of prednisone 59 Kanigicherla D, Gummadova J, McKenzie EA, et al. Anti-PLA2R
and ACE inhibitor versus ACE-inhibitor therapy alone in patients antibodies measured by ELISA predict long-term outcome in a
with IgA nephropathy: a randomized controlled trial. prevalent population of patients with idiopathic membranous
Am J Kidney Dis 2009; 53: 26–32. nephropathy. Kidney Int 2013; 83: 940–48.
38 Manno C, Torres DD, Rossini M, Pesce F, Schena FP. Randomized 60 Hoxha E, Thiele I, Zahner G, Panzer U, Harendza S, Stahl RA.
controlled clinical trial of corticosteroids plus ACE-inhibitors with Phospholipase A2 receptor autoantibodies and clinical outcome in
long-term follow-up in proteinuric IgA nephropathy. patients with primary membranous nephropathy. J Am Soc Nephrol
Nephrol Dial Transplant 2009; 24: 3694–701. 2014; 25: 1357–66.
39 Lv J, Xu D, Perkovic V, et al. Corticosteroid therapy in IgA 61 Ruggenenti P, Debiec H, Ruggiero B, et al. Anti-phospholipase A2
nephropathy. J Am Soc Nephrol 2012; 23: 1108–16. receptor antibody titer predicts post-rituximab outcome of
40 Eitner F, Ackermann D, Hilgers RD, Floege J. Supportive versus membranous nephropathy. J Am Soc Nephrol 2015; 26: 2545–58.
immunosuppressive therapy of progressive IgA nephropathy 62 Hofstra JM, Beck LH Jr, Beck DM, Wetzels JF, Salant DJ.
(STOP) IgAN trial: rationale and study protocol. J Nephrol 2008; Anti-phospholipase A receptor antibodies correlate with clinical
21: 284–89. status in idiopathic membranous nephropathy.
41 Rauen T, Eitner F, Fitzner C, et al. Intensive supportive care plus Clin J Am Soc Nephrol 2011; 6: 1286–91.
immunosuppression in IgA nephropathy. N Engl J Med 2015; 63 Bech AP, Hofstra JM, Brenchley PE, Wetzels JF. Association of
373: 2225–36. anti-PLA(2)R antibodies with outcomes after immunosuppressive
42 Tang SC, Tang AW, Wong SS, Leung JC, Ho YW, Lai KN. Long-term therapy in idiopathic membranous nephropathy.
study of mycophenolate mofetil treatment in IgA nephropathy. Clin J Am Soc Nephrol 2014; 9: 1386–92.
Kidney Int 2010; 77: 543–49. 64 Kattah A, Ayalon R, Beck LH Jr, et al. Anti-phospholipase A(2)
43 Maes BD, Oyen R, Claes K, et al. Mycophenolate mofetil in IgA receptor antibodies in recurrent membranous nephropathy.
nephropathy: results of a 3-year prospective placebo-controlled Am J Transplant 2015; 15: 1349–59.
randomized study. Kidney Int 2004; 65: 1842–49. 65 Beck LH Jr, Fervenza FC, Beck DM, et al. Rituximab-induced
44 Frisch G, Lin J, Rosenstock J, et al. Mycophenolate mofetil (MMF) depletion of anti-PLA2R autoantibodies predicts response in
vs placebo in patients with moderately advanced IgA nephropathy: membranous nephropathy. J Am Soc Nephrol 2011; 22: 1543–50.
a double-blind randomized controlled trial. Nephrol Dial Transplant 66 Hoxha E, Harendza S, Pinnschmidt HO, et al. Spontaneous
2005; 20: 2139–45. remission of proteinuria is a frequent event in phospholipase A2
45 Hogg RJ, Bay RC, Jennette JC, et al. Randomized controlled trial of receptor antibody negative patients with membranous nephropathy.
mycophenolate mofetil in children, adolescents, and adults with Nephrol Dial Transplant 2015; 30: 1862–69.
IgA nephropathy. Am J Kidney Dis 2015; 66: 783–91. 67 Polanco N, Gutierrez E, Covarsi A, et al. Spontaneous remission of
46 Lv J, Zhang H, Cui Z, Su T, Zhang Y, Wang H. Delayed severe nephrotic syndrome in idiopathic membranous nephropathy.
pneumonia in mycophenolate mofetil-treated patients with IgA J Am Soc Nephrol 2010; 21: 697–704.
nephropathy. Nephrol Dial Transplant 2008; 23: 2868–72. 68 Hofstra JM, Branten AJ, Wirtz JJ, Noordzij TC, du Buf-Vereijken PW,
47 Pozzi C, Andrulli S, Pani A, et al. Addition of azathioprine to Wetzels JF. Early versus late start of immunosuppressive therapy in
corticosteroids does not benefit patients with IgA nephropathy. idiopathic membranous nephropathy: a randomized controlled trial.
J Am Soc Nephrol 2010; 21: 1783–90. Nephrol Dial Transplant 2010; 25: 129–36.
48 Smerud HK, Fellstrom B, Hallgren R, Osagie S, Venge P, 69 van den Brand JA, van Dijk PR, Hofstra JM, Wetzels JF.
Kristjansson G. Gluten sensitivity in patients with IgA nephropathy. Long-term outcomes in idiopathic membranous nephropathy
Nephrol Dial Transplant 2009; 24: 2476–81. using a restrictive treatment strategy. J Am Soc Nephrol 2014;
49 Smerud HK, Barany P, Lindstrom K, et al. New treatment for IgA 25: 150–58.
nephropathy: enteric budesonide targeted to the ileocecal region 70 Howman A, Chapman TL, Langdon MM, et al.
ameliorates proteinuria. Nephrol Dial Transplant 2011; Immunosuppression for progressive membranous nephropathy:
26: 3237–42. a UK randomised controlled trial. Lancet 2013; 381: 744–51.
50 Kawamura T, Yoshimura M, Miyazaki Y, et al, for the Special IgA 71 Dussol B, Morange S, Burtey S, et al. Mycophenolate mofetil
Nephropathy Study Group. A multicenter randomized controlled monotherapy in membranous nephropathy: a 1-year randomized
trial of tonsillectomy combined with steroid pulse therapy in controlled trial. Am J Kidney Dis 2008; 52: 699–705.
patients with immunoglobulin A nephropathy. 72 Chen Y, Schieppati A, Cai G, et al. Immunosuppression for
Nephrol Dial Transplant 2014; 29: 1546–53. membranous nephropathy: a systematic review and meta-analysis
51 Floege J, Feehally J. The mucosa-kidney axis in IgA nephropathy. of 36 clinical trials. Clin J Am Soc Nephrol 2013; 8: 787–96.
Nat Rev Nephrol 2015; published online Dec 30. DOI:10.1038/ 73 Ruggenenti P, Cravedi P, Chianca A, et al. Rituximab in idiopathic
nrneph.2015.208. membranous nephropathy. J Am Soc Nephrol 2012; 23: 1416–25.
52 Leeaphorn N, Kue APP, Thamcharoen N, Ungprasert P, Stokes MB, 74 Hoxha E, Thiele I, Zahner G, Panzer U, Harendza S, Stahl RA.
Knight EL. Prevalence of cancer in membranous nephropathy: Phospholipase A2 receptor autoantibodies and clinical outcome in
a systematic review and meta-analysis of observational studies. patients with primary membranous nephropathy. J Am Soc Nephrol
Am J Nephrol 2014; 40: 29–35. 2014; 25: 1357–66.
53 Stanescu HC, Arcos-Burgos M, Medlar A, et al. Risk HLA-DQA1 75 Herlitz LC, Bomback AS, Stokes MB, Radhakrishnan J, D’Agati VD,
and PLA(2)R1 alleles in idiopathic membranous nephropathy. Markowitz GS. IgA nephropathy with minimal change disease.
N Engl J Med 2011; 364: 616–26. Clin J Am Soc Nephrol 2014; 9: 1033–39.
54 Fresquet M, Jowitt TA, Gummadova J, et al. Identification of a 76 Shankland SJ, Smeets B, Pippin JW, Moeller MJ. The emergence of
major epitope recognized by PLA2R autoantibodies in primary the glomerular parietal epithelial cell. Nat Rev Nephrol 2014; 10: 158–73.
membranous nephropathy. J Am Soc Nephrol 2015; 26: 302–13. 77 Hakroush S, Cebulla A, Schaldecker T, Behr D, Mundel P, Weins A.
55 Kao L, Lam V, Waldman M, Glassock RJ, Zhu Q. Identification of Extensive podocyte loss triggers a rapid parietal epithelial cell
the immunodominant epitope region in phospholipase A2 response. J Am Soc Nephrol 2014; 25: 927–38.
receptor-mediating autoantibody binding in idiopathic 78 Smeets B, Stucker F, Wetzels J, et al. Detection of activated parietal
membranous nephropathy. J Am Soc Nephrol 2015; 26: 291–301. epithelial cells on the glomerular tuft distinguishes early focal
56 Tomas NM, Beck LH Jr, Meyer-Schwesinger C, et al. segmental glomerulosclerosis from minimal change disease.
Thrombospondin type-1 domain-containing 7A in idiopathic Am J Pathol 2014; 184: 3239–48.
membranous nephropathy. N Engl J Med 2014; 371: 2277–87. 79 Chugh SS, Clement LC, Mace C. New insights into human minimal
57 Debiec H, Lefeu F, Kemper MJ, et al. Early-childhood membranous change disease: lessons from animal models. Am J Kidney Dis 2012;
nephropathy due to cationic bovine serum albumin. N Engl J Med 59: 284–92.
2011; 364: 2101–10. 80 Herlitz LC, Markowitz GS, Farris AB, et al. Development of focal
58 Du Y, Li J, He F, et al. The diagnosis accuracy of PLA2R-AB in the segmental glomerulosclerosis after anabolic steroid abuse.
diagnosis of idiopathic membranous nephropathy: a meta-analysis. J Am Soc Nephrol 2010; 21: 163–72.
PLoS One 2014; 9: e104936.
2046 www.thelancet.com Vol 387 May 14, 2016
Seminar
81 Clement LC, Avila-Casado C, Mace C, et al. Podocyte-secreted 104 Li X, Xu N, Li H, et al. Tacrolimus as rescue therapy for
angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive adult-onset refractory minimal change nephrotic syndrome with
nephrotic syndrome. Nat Med 2011; 17: 117–22. reversible acute renal failure. Nephrol Dial Transplant 2013;
82 Wei C, El Hindi S, Li J, et al. Circulating urokinase receptor as a 28: 2306–12.
cause of focal segmental glomerulosclerosis. Nat Med 2011; 105 Choudhry S, Bagga A, Hari P, Sharma S, Kalaivani M, Dinda A.
17: 952–60. Efficacy and safety of tacrolimus versus cyclosporine in children
83 McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors with steroid-resistant nephrotic syndrome: a randomized controlled
in idiopathic nephrotic syndrome and focal segmental trial. Am J Kidney Dis 2009; 53: 760–69.
glomerulosclerosis. Clin J Am Soc Nephrol 2010; 5: 2115–21. 106 Braun N, Schmutzler F, Lange C, et al. Immunosuppressive
84 Maas RJ, Deegens JK, Wetzels JF. Permeability factors in idiopathic treatment for focal segmental glomerulosclerosis in adults.
nephrotic syndrome: historical perspectives and lessons for the Cochrane Database Syst Rev 2008; 3: CD003233.
future. Nephrol Dial Transplant 2014; 29: 2207–16. 107 Iijima K, Sako M, Nozu K, et al, for the Rituximab for
85 Cathelin D, Placier S, Ploug M, et al. Administration of Childhood-onset Refractory Nephrotic Syndrome (RCRNS) Study
recombinant soluble urokinase receptor per se is not sufficient to Group. Rituximab for childhood-onset, complicated, frequently
induce podocyte alterations and proteinuria in mice. relapsing nephrotic syndrome or steroid-dependent nephrotic
J Am Soc Nephrol 2014; 25: 1662–68. syndrome: a multicentre, double-blind, randomised,
86 Bock ME, Price HE, Gallon L, Langman CB. Serum soluble placebo-controlled trial. Lancet 2014; 384: 1273–81.
urokinase-type plasminogen activator receptor levels and idiopathic 108 Ravani P, Magnasco A, Edefonti A, et al. Short-term effects of
FSGS in children: a single-center report. Clin J Am Soc Nephrol rituximab in children with steroid- and calcineurin-dependent
2013; 8: 1304–11. nephrotic syndrome: a randomized controlled trial.
87 Huang J, Liu G, Zhang YM, et al. Plasma soluble urokinase receptor Clin J Am Soc Nephrol 2011; 6: 1308–15.
levels are increased but do not distinguish primary from secondary 109 Gulati A, Sinha A, Jordan SC, et al. Efficacy and safety of treatment
focal segmental glomerulosclerosis. Kidney Int 2013; 84: 366–72. with rituximab for difficult steroid-resistant and -dependent
88 Meijers B, Maas RJ, Sprangers B, et al. The soluble urokinase nephrotic syndrome: multicentric report. Clin J Am Soc Nephrol
receptor is not a clinical marker for focal segmental 2010; 5: 2207–12.
glomerulosclerosis. Kidney Int 2014; 85: 636–40. 110 Munyentwali H, Bouachi K, Audard V, et al. Rituximab is an
89 Sinha A, Bajpai J, Saini S, et al. Serum-soluble urokinase receptor efficient and safe treatment in adults with steroid-dependent
levels do not distinguish focal segmental glomerulosclerosis from minimal change disease. Kidney Int 2013; 83: 511–16.
other causes of nephrotic syndrome in children. Kidney Int 2014; 111 Ruggenenti P, Ruggiero B, Cravedi P, et al, for the Rituximab in
85: 649–58. Nephrotic Syndrome of Steroid-Dependent or Frequently Relapsing
90 Wada T, Nangaku M, Maruyama S, et al. A multicenter cross-sectional Minimal Change Disease Or Focal Segmental Glomerulosclerosis
study of circulating soluble urokinase receptor in Japanese patients (NEMO) Study Group. Rituximab in steroid-dependent or
with glomerular disease. Kidney Int 2014; 85: 641–48. frequently relapsing idiopathic nephrotic syndrome.
J Am Soc Nephrol 2014; 25: 850–63.
91 Garin EH, Mu W, Arthur JM, et al. Urinary CD80 is elevated in
minimal change disease but not in focal segmental 112 Fornoni A, Sageshima J, Wei C, et al. Rituximab targets podocytes
glomerulosclerosis. Kidney Int 2010; 78: 296–302. in recurrent focal segmental glomerulosclerosis. Sci Transl Med
2011; 3: 85ra46.
92 Laurin LP, Lu M, Mottl AK, Blyth ER, Poulton CJ, Weck KE.
Podocyte-associated gene mutation screening in a heterogeneous 113 Basu B. Ofatumumab for rituximab-resistant nephrotic syndrome.
cohort of patients with sporadic focal segmental glomerulosclerosis. N Engl J Med 2014; 370: 1268–70.
Nephrol Dial Transplant 2014; 29: 2062–69. 114 Yu CC, Fornoni A, Weins A, et al. Abatacept in B7-1-positive
93 Sadowski CE, Lovric S, Ashraf S, et al, for the SRNS Study Group. proteinuric kidney disease. N Engl J Med 2013; 369: 2416–23.
A single-gene cause in 29.5% of cases of steroid-resistant nephrotic 115 Benigni A, Gagliardini E, Remuzzi G. Abatacept in B7-1-positive
syndrome. J Am Soc Nephrol 2015; 26: 1279–89. proteinuric kidney disease. N Engl J Med 2014; 370: 1261–63.
94 Gast C, Pengelly RJ, Lyon M, et al. Collagen (COL4A) mutations are 116 Garin EH, Reiser J, Cara-Fuentes G, et al. Case series: CTLA4-IgG1
the most frequent mutations underlying adult focal segmental therapy in minimal change disease and focal segmental
glomerulosclerosis. Nephrol Dial Transplant 2015; published online glomerulosclerosis. Pediatr Nephrol 2015; 30: 469–77.
Sept 7. DOI:10.1093/ndt/gfv325. 117 Trachtman H, Vento S, Herreshoff E, et al. Efficacy of galactose and
95 Barua M, Shieh E, Schlondorff J, Genovese G, Kaplan BS, adalimumab in patients with resistant focal segmental
Pollak MR. Exome sequencing and in vitro studies identified glomerulosclerosis: report of the font clinical trial group.
podocalyxin as a candidate gene for focal and segmental BMC Nephrol 2015; 16: 111.
glomerulosclerosis. Kidney Int 2014; 85: 124–33. 118 Fervenza FC, Sethi S, Glassock RJ. Idiopathic
96 Barua M, Stellacci E, Stella L, et al. Mutations in PAX2 associate membranoproliferative glomerulonephritis: does it exist?
with adult-onset FSGS. J Am Soc Nephrol 2014; 25: 1942–53. Nephrol Dial Transplant 2012; 27: 4288–94.
97 Cong EH, Bizet AA, Boyer O, et al. A homozygous missense 119 Sethi S, Zand L, Leung N, et al. Membranoproliferative
mutation in the ciliary gene TTC21B causes familial FSGS. glomerulonephritis secondary to monoclonal gammopathy.
J Am Soc Nephrol 2014; 25: 2435–43. Clin J Am Soc Nephrol 2010; 5: 770–82.
98 Gbadegesin RA, Hall G, Adeyemo A, et al. Mutations in the gene 120 Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis:
that encodes the F-actin binding protein anillin cause FSGS. pathogenetic heterogeneity and proposal for a new classification.
J Am Soc Nephrol 2014; 25: 1991–2002. Semin Nephrol 2011; 31: 341–48.
99 Hall G, Gbadegesin RA, Lavin P, et al. A novel missense mutation 121 Abrera-Abeleda MA, Nishimura C, Frees K, et al. Allelic variants of
of Wilms’ tumor 1 causes autosomal dominant FSGS. complement genes associated with dense deposit disease.
J Am Soc Nephrol 2015; 26: 831–43. J Am Soc Nephrol 2011; 22: 1551–59.
100 Korkmaz E, Lipska-Zietkiewicz BS, Boyer O, et al. ADCK4-associated 122 Gale DP, de Jorge EG, Cook HT, et al. Identification of a mutation
glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol in complement factor H-related protein 5 in patients of Cypriot
2016; 27: 63–68. origin with glomerulonephritis. Lancet 2010; 376: 794–801.
101 Li L, Zhang T, Diao W, et al. Role of myeloid-derived suppressor 123 Sethi S, Fervenza FC, Zhang Y, et al. Proliferative
cells in glucocorticoid-mediated amelioration of FSGS. glomerulonephritis secondary to dysfunction of the alternative
J Am Soc Nephrol 2015; 26: 2183–97. pathway of complement. Clin J Am Soc Nephrol 2011; 6: 1009–17.
102 Gipson DS, Trachtman H, Kaskel FJ, et al. Clinical trial of focal 124 Sethi S, Nester CM, Smith RJ. Membranoproliferative
segmental glomerulosclerosis in children and young adults. glomerulonephritis and C3 glomerulopathy: resolving the
Kidney Int 2011; 80: 868–78. confusion. Kidney Int 2012; 81: 434–41.
103 Deegens JK, Wetzels JF. Immunosuppressive treatment of focal 125 Zhang Y, Meyer NC, Wang K, et al. Causes of alternative pathway
segmental glomerulosclerosis: lessons from a randomized dysregulation in dense deposit disease. Clin J Am Soc Nephrol 2012;
controlled trial. Kidney Int 2011; 80: 798–801. 7: 265–74.
www.thelancet.com Vol 387 May 14, 2016 2047
Seminar
126 Medjeral-Thomas NR, O’Shaughnessy MM, O’Regan JA, et al. 138 Ohlsson S, Herlitz H, Lundberg S, et al. Circulating anti-glomerular
C3 glomerulopathy: clinicopathologic features and predictors of basement membrane antibodies with predominance of subclass
outcome. Clin J Am Soc Nephrol 2014; 9: 46–53. IgG4 and false-negative immunoassay test results in anti-
127 Bu F, Borsa NG, Jones MB, et al. High-throughput genetic testing glomerular basement membrane disease. Am J Kidney Dis 2014;
for thrombotic microangiopathies and C3 glomerulopathies. 63: 289–93.
J Am Soc Nephrol 2015; published online Aug 17. DOI:10.1681/ 139 Jia XY, Hu SY, Chen JL, et al. The clinical and immunological
ASN.2015040385. features of patients with combined anti-glomerular basement
128 Zand L, Lorenz EC, Cosio FG, et al. Clinical findings, pathology, membrane disease and membranous nephropathy. Kidney Int 2014;
and outcomes of C3GN after kidney transplantation. 85: 945–52.
J Am Soc Nephrol 2014; 25: 1110–17. 140 Cui Z, Zhao J, Jia XY, et al. Anti-glomerular basement membrane
129 Bomback AS, Smith RJ, Barile GR, et al. Eculizumab for dense disease: outcomes of different therapeutic regimens in a large
deposit disease and C3 glomerulonephritis. Clin J Am Soc Nephrol single-center Chinese cohort study. Medicine (Baltimore) 2011;
2012; 7: 748–56. 90: 303–11.
130 Daina E, Noris M, Remuzzi G. Eculizumab in a patient with 141 Touzot M, Poisson J, Faguer S, et al. Rituximab in anti-GBM
dense-deposit disease. N Engl J Med 2012; 366: 1161–63. disease: a retrospective study of 8 patients. J Autoimmun 2015;
131 Herlitz LC, Bomback AS, Markowitz GS, et al. Pathology after 60: 74–79.
eculizumab in dense deposit disease and C3 GN. J Am Soc Nephrol 142 Tang W, McDonald SP, Hawley CM, et al. Anti-glomerular
2012; 23: 1229–37. basement membrane antibody disease is an uncommon cause of
132 Chen Q, Muller D, Rudolph B, et al. Combined C3b and factor B end-stage renal disease. Kidney Int 2013; 83: 503–10.
autoantibodies and MPGN type II. N Engl J Med 2011; 365: 2340–42. 143 Vizjak A, Ferluga D, Rozic M, et al. Pathology, clinical
133 Chen Q, Wiesener M, Eberhardt HU, et al. Complement factor presentations, and outcomes of C1q nephropathy. J Am Soc Nephrol
H-related hybrid protein deregulates complement in dense deposit 2008; 19: 2237–44.
disease. J Clin Invest 2014; 124: 145–55. 144 Nasr SH, Valeri AM, Cornell LD, et al. Fibrillary
134 Rabasco C, Cavero T, Roman E, et al. Effectiveness of glomerulonephritis: a report of 66 cases from a single institution.
mycophenolate mofetil in C3 glomerulonephritis. Kidney Int 2015; Clin J Am Soc Nephrol 2011; 6: 775–84.
88: 1153–60. 145 Javaugue V, Karras A, Glowacki F, et al. Long-term kidney disease
135 Pedchenko V, Bondar O, Fogo AB, et al. Molecular architecture of outcomes in fibrillary glomerulonephritis: a case series of
the Goodpasture autoantigen in anti-GBM nephritis. N Engl J Med 27 patients. Am J Kidney Dis 2013; 62: 679–90.
2010; 363: 343–54. 146 Palmer SC, Sciancalepore M, Strippoli GF. Trial quality in
136 Zhao J, Cui Z, Yang R, Jia XY, Zhang Y, Zhao MH. Anti-glomerular nephrology: how are we measuring up? Am J Kidney Dis 2011;
basement membrane autoantibodies against different target 58: 335–37.
antigens are associated with disease severity. Kidney Int 2009;
76: 1108–15.
137 Cui Z, Zhao J, Jia XY, Zhu SN, Zhao MH. Clinical features and
outcomes of anti-glomerular basement membrane disease in older
patients. Am J Kidney Dis 2011; 57: 575–82.
2048 www.thelancet.com Vol 387 May 14, 2016
You might also like
- 7 - Pile Cutting & HackingDocument2 pages7 - Pile Cutting & HackingRafee Pie93% (14)
- Nephrotic Syndrome: BackgroundDocument25 pagesNephrotic Syndrome: BackgroundsDamnNo ratings yet
- Nephrotic Vs Nephritic SyndromeDocument80 pagesNephrotic Vs Nephritic Syndromevan016_bunnyNo ratings yet
- READ How A Common Soup ThickenerDocument4 pagesREAD How A Common Soup ThickenerOgooNkem100% (1)
- A Apb 2017 On Site ProgramDocument68 pagesA Apb 2017 On Site Programjoelmuk2001No ratings yet
- Nephritic SyndromeDocument24 pagesNephritic SyndromeMuhamed Al Rohani100% (1)
- GlomerulonephritisDocument58 pagesGlomerulonephritisJosa Anggi Pratama0% (1)
- File 128 - Zohaib Biopsy Marked4!6!2019Document6 pagesFile 128 - Zohaib Biopsy Marked4!6!2019nreena aslamNo ratings yet
- NEPHRITISDocument37 pagesNEPHRITISJay RathvaNo ratings yet
- Emergency Medicine ST4Document7 pagesEmergency Medicine ST4QusaiBadrNo ratings yet
- Glomerulonephritis: Lecturer Prof. Yu.R. KovalevDocument39 pagesGlomerulonephritis: Lecturer Prof. Yu.R. Kovalevalfaz lakhani100% (1)
- Bishop ScoreDocument3 pagesBishop ScoreAlain SánchezNo ratings yet
- Acute GlomerulonephritisDocument4 pagesAcute GlomerulonephritisJulliza Joy PandiNo ratings yet
- Seminar On Nephrotic Syndrome: Medical Surgical NursingDocument15 pagesSeminar On Nephrotic Syndrome: Medical Surgical NursingGargi MP100% (1)
- XLR8 2.0Document2 pagesXLR8 2.0EvgeniyNo ratings yet
- Approach To HematuriaDocument45 pagesApproach To HematuriaArun GeorgeNo ratings yet
- Practice Exam - HematologyDocument2 pagesPractice Exam - HematologyrlinaoNo ratings yet
- Rapidly Progressive GlomerulonephritisDocument17 pagesRapidly Progressive GlomerulonephritisEasyOrientDNo ratings yet
- Management of Patients With Nephrotic Syndrome: Sophie de Seigneux, Pierre-Yves MartinDocument7 pagesManagement of Patients With Nephrotic Syndrome: Sophie de Seigneux, Pierre-Yves MartinGarit Hapsari KiranaNo ratings yet
- Post Streptococus GNDocument14 pagesPost Streptococus GNRahma Cita HalidaNo ratings yet
- 10 Primary Glumerulopathies III - GKDocument2 pages10 Primary Glumerulopathies III - GKGerarld Immanuel KairupanNo ratings yet
- Acute GlomerulonephritisDocument18 pagesAcute GlomerulonephritisdanielaNo ratings yet
- Gnaps EmedicineDocument13 pagesGnaps Emedicineharyanti lupitaNo ratings yet
- Clinical Features and Diagnosis of Small-Vessel VasculitisDocument13 pagesClinical Features and Diagnosis of Small-Vessel VasculitissigmundmaharajanNo ratings yet
- TB, Review, PathoDocument4 pagesTB, Review, PathoKrizzia LaturnasNo ratings yet
- Lecture 4 (1of3) - Nephritic SyndromeDocument45 pagesLecture 4 (1of3) - Nephritic SyndromeAliye BaramNo ratings yet
- Nephrotic Syndrome: DR Thuvaraka WareDocument5 pagesNephrotic Syndrome: DR Thuvaraka Warechloe1411No ratings yet
- Acute Poststreptococcal Glomerulonephritis (Apsgn) in ChildrenDocument22 pagesAcute Poststreptococcal Glomerulonephritis (Apsgn) in ChildrenOlivia PutriNo ratings yet
- Acute GlomerulonephritisDocument32 pagesAcute GlomerulonephritisEzekiel moraraNo ratings yet
- Acute GlomerulonephritisDocument28 pagesAcute GlomerulonephritisPaul SinsNo ratings yet
- GlomerulonephritisDocument35 pagesGlomerulonephritisapi-19916399No ratings yet
- Isolated Glomerular Disease With Recurrent Gross HematuriaDocument17 pagesIsolated Glomerular Disease With Recurrent Gross HematuriaArun GeorgeNo ratings yet
- What Is Glomerulonephritis?Document7 pagesWhat Is Glomerulonephritis?SSNo ratings yet
- Agn PDFDocument6 pagesAgn PDFMohamed ZiadaNo ratings yet
- Nephritic Syndrome - StatPearls - NCBI BookshelfDocument10 pagesNephritic Syndrome - StatPearls - NCBI Bookshelfraul.villarrealNo ratings yet
- Gambaran Klinis Glomerulonefritis Akut Pada Anak Di RSUP Prof. Dr. R. D. Kandou ManadoDocument5 pagesGambaran Klinis Glomerulonefritis Akut Pada Anak Di RSUP Prof. Dr. R. D. Kandou Manadocleria riceNo ratings yet
- What Is Acute Glomerulonephritis?: Acute Glomerulonephritis (GN) Comprises A Specific Set of Renal Diseases inDocument6 pagesWhat Is Acute Glomerulonephritis?: Acute Glomerulonephritis (GN) Comprises A Specific Set of Renal Diseases inAnnapoorna SHNo ratings yet
- MANUSCRIPTDocument11 pagesMANUSCRIPTANA DelafuenteNo ratings yet
- Nephrotic Syndrome: Signs and SymptomsDocument7 pagesNephrotic Syndrome: Signs and SymptomsErika ManubayNo ratings yet
- APSGNDocument3 pagesAPSGNmadimadi11No ratings yet
- Glomerular DiseasesDocument28 pagesGlomerular DiseasesNagaraj Reddy100% (1)
- Nephrotic Syndrome in Children-LectureDocument52 pagesNephrotic Syndrome in Children-LectureLubinda SitaliNo ratings yet
- GlomerulonephritisDocument46 pagesGlomerulonephritisvanessaNo ratings yet
- ASPGNDocument17 pagesASPGNAdLi DLi DLiNo ratings yet
- Lecture Note On Renal Diseases For Medical Students - GNDocument10 pagesLecture Note On Renal Diseases For Medical Students - GNEsayas KebedeNo ratings yet
- Acute Poststreptococcal Glomerulonephritis An UpdateDocument6 pagesAcute Poststreptococcal Glomerulonephritis An UpdateJuan Antonio Herrera LealNo ratings yet
- Glomerulonefritis ComplementoDocument10 pagesGlomerulonefritis ComplementoPaola ZuluagaNo ratings yet
- Table 163-1: Glomerulonephritis (GN) - Poststreptococcal GlomerulonephritisDocument6 pagesTable 163-1: Glomerulonephritis (GN) - Poststreptococcal GlomerulonephritisAndi Riza SyafitriNo ratings yet
- Overview of Heavy Proteinuria and The Nephrotic Syndrome - UpToDateDocument37 pagesOverview of Heavy Proteinuria and The Nephrotic Syndrome - UpToDateAdrian MoranNo ratings yet
- PSGNDocument16 pagesPSGNArun GeorgeNo ratings yet
- Review Article: An Approach To The Child With Acute GlomerulonephritisDocument4 pagesReview Article: An Approach To The Child With Acute GlomerulonephritisLu Jordy LuhurNo ratings yet
- Superior Vena Cava SyndromeDocument16 pagesSuperior Vena Cava SyndromeHUGO DANIEL VAZQUEZ GOMEZNo ratings yet
- Emergent Management of Acute Glomerulonephritis - Overview, Emergency Department Care, ConsultationsDocument4 pagesEmergent Management of Acute Glomerulonephritis - Overview, Emergency Department Care, ConsultationsHafiz Maulana AhmadNo ratings yet
- Recent Update in The Management of Childhood Nephr PDFDocument8 pagesRecent Update in The Management of Childhood Nephr PDFREHNUMA URMINo ratings yet
- Glomerulonephritis Acute UKDocument12 pagesGlomerulonephritis Acute UKmadimadi11No ratings yet
- Nephritic SyndromeDocument19 pagesNephritic SyndromesangheetaNo ratings yet
- Primary Glomerulonephritis UG LectureDocument50 pagesPrimary Glomerulonephritis UG LectureMalik Mohammad AzharuddinNo ratings yet
- Acute GlomerulonephritisDocument27 pagesAcute GlomerulonephritisKumara GuruNo ratings yet
- Acute Glomerulonephritis PDFDocument27 pagesAcute Glomerulonephritis PDFRaj RNo ratings yet
- Chronic GoutyDocument4 pagesChronic GoutyDavid Lance MarquezNo ratings yet
- Kidney Research and Clinical Practice: Jürgen FloegeDocument8 pagesKidney Research and Clinical Practice: Jürgen FloegeAnita PrastiwiNo ratings yet
- Predictors of Prognosis in Iga Nephropathy: Review ArticleDocument4 pagesPredictors of Prognosis in Iga Nephropathy: Review ArticledavidpuaNo ratings yet
- Letters To The Editor: Membranous Glomerulonephritis in A Patient With SyphilisDocument2 pagesLetters To The Editor: Membranous Glomerulonephritis in A Patient With SyphilisBetty Romero BarriosNo ratings yet
- CE Update: Wegener's Granulomatosis: A Review of The Clinical Implications, Diagnosis, and TreatmentDocument3 pagesCE Update: Wegener's Granulomatosis: A Review of The Clinical Implications, Diagnosis, and TreatmentAfif FaiziNo ratings yet
- Lecture (5) - RPGN - CKDDocument20 pagesLecture (5) - RPGN - CKDpoojabappanna25No ratings yet
- Heart Valve Disease: State of the ArtFrom EverandHeart Valve Disease: State of the ArtJose ZamoranoNo ratings yet
- Final Case Study 2Document24 pagesFinal Case Study 2api-351727746100% (1)
- Answer To The Question No. 1/ (A)Document3 pagesAnswer To The Question No. 1/ (A)zahadul AlamNo ratings yet
- Oxfam Minimum Requirements For WASH Programmes PDFDocument56 pagesOxfam Minimum Requirements For WASH Programmes PDFLinel DzawoNo ratings yet
- Food Preparation Evaluation Guide: Olivarez College Tagaytay College of NursingDocument2 pagesFood Preparation Evaluation Guide: Olivarez College Tagaytay College of NursingRaquel M. MendozaNo ratings yet
- Acanthosis NigricansDocument4 pagesAcanthosis Nigricansilham raymanaNo ratings yet
- Role of Private Sectors in Health Service and ManagementDocument2 pagesRole of Private Sectors in Health Service and Managementcopy smartNo ratings yet
- Atos CFSDocument22 pagesAtos CFSPaul SmithNo ratings yet
- Component of Physical FitnessDocument16 pagesComponent of Physical FitnessMYRA BACSAFRANo ratings yet
- Role of Ngos in Rural Development in IndiaDocument17 pagesRole of Ngos in Rural Development in IndiaRavia SharmaNo ratings yet
- Revised ToR For OH Project Baseline Evaluation 1Document6 pagesRevised ToR For OH Project Baseline Evaluation 1amosmutyabaNo ratings yet
- Android Projects - 1000 ProjectsDocument14 pagesAndroid Projects - 1000 ProjectsFassy ur RehmanNo ratings yet
- HSR2 5 E649Document9 pagesHSR2 5 E649Samsul BahriNo ratings yet
- NCM 101 Rooreading To The Max Aug. 13, 2018Document8 pagesNCM 101 Rooreading To The Max Aug. 13, 2018rimeoznekNo ratings yet
- Metabolic Dysregulation: America's Got Side EffectsDocument28 pagesMetabolic Dysregulation: America's Got Side EffectsNaiana PaulaNo ratings yet
- Private Hospital Sector Development: An Exploratory Study On Providers Perspective in Addis Ababa, EthiopiaDocument7 pagesPrivate Hospital Sector Development: An Exploratory Study On Providers Perspective in Addis Ababa, EthiopiaEDENNo ratings yet
- Hydraulic Oil 68: Safety Data SheetDocument5 pagesHydraulic Oil 68: Safety Data Sheetjerry dauteyNo ratings yet
- Worst Food For HealthDocument2 pagesWorst Food For HealthZhenya JNo ratings yet
- Soal Jawaban Uts Bahasa Inggris 2 2020Document5 pagesSoal Jawaban Uts Bahasa Inggris 2 2020Luki RaKiraNo ratings yet
- DOFA Plastic SurgeryDocument2 pagesDOFA Plastic SurgeryHarol EscalanteNo ratings yet
- Building EnvironmentDocument11 pagesBuilding Environmentserena KazokuNo ratings yet
- Ethical Implications of Research ActivityDocument3 pagesEthical Implications of Research ActivitymothicyNo ratings yet
- NRG305 Midterm ReviewerDocument22 pagesNRG305 Midterm ReviewerEMMY FLOR VALMORIANo ratings yet