ese Che Sada, 197 387-391 pel 9 dee Chin Sona 197
Tre ahs ned an
ccrteaica SEANDINAVICA
Crystal Structure of the Double Carbonate K,Caz(CO3)3
Camilla Winbo,* Dan Bostrém** and Matthias Gébbels”
“Department of Inorganic Chemistry, Umea University, $-901 87 Umed, Sweden and "Institut far Kristallographie,
RWTH Aachen, D-52056 Aachen, Germany
Winbo, C., BostrOm, D. and Gabbels, M., 1997. Crystal Structure ofthe Double
Carbonate K;Cas{CO,), ~ Acta Chem, Seand. 51: 387-391. © Acta Chemica
Scandinavica 1997,
‘The crystal structure of K,Ca,(CO,), was determined from single-crystal
X-ray difracton data. This double carbonate erytalises in the spsce BrouP
B3 (No 146). with unitoed dimensions «= 15010(4), = 8615) Ay
12639(9) AY and 2 ~6. Anisotropic refinements gave Ry ~ 0.018 for 816 snigue
refections. The two eystallogaphicaly ferent carbonate ions are located in
Colgrans around the thce-fold sacs at which 1 of the cations ae stated (one
‘Ca** and one K"), the remainder being located in general positions between
the carbonate coluris,‘The carbouats ona are more or lew ocned eat
th (001 plane and donot form an unequivocal layered stactre Experimental
and ealcoted powder Xray cifraction date for KyCay(CO,), are presented
‘The first published investigation of the K,CO,-CaCO,
system was performed by Nigali? in 1916, Niggli studied
the KCOy-rich side of the system, in I atm of CO, and
in the temperature range 700-900°C, and found one
intermediate phase; K:Ca(COs):, melting at 813°C.
Kroger et al? investigated the phase relations and pre-
sented a phase diagram for the binary system KCO,-
CaCO, in the temperature range 500-1400°C, at
approximately 50>bar. They found two intermediate
phases, the double carbonates K,Ca(CO,); and
K,Cax(CO,),. Walenta® describes the occurrence of a
new carbonate mineral containing calcium and potassium
as main cation components. From optical crystallog-
raphy, Walenta concluded that this new mineral belongs
to the ‘rhombohedral system’. Cooper et al.‘ determined
the phase relations in the ternary system NaCO,-
K,CO,-CaCOy at kbar. In the binary sub-system
K3CO,-CaC0O, they describe three intermediate phases,
two K,Ca(CO,), phases, fairchildite and btscli
(hich is low temperature modification of the former)
and an unnamed phase, K,Ca,(CO,),, referred to a8
phase A. K,Ca,(CO,), is an incongruently melting phase
(810°C) which decomposes at 512°C to calcite and
batschite. The erystals of phase A are described as pale
yellow-green granules, with grains that always exhibit
polysynthetic twin lamellae parallel and perpendicular to
the c-axis.
Although this double carbonate KCa,(COs), has
been known since 1943, the crystal structure has not
been determined; in fact not even an unambiguous
powder diffraction pattern is available, The two other
"To whom correspondence should be addressed.
(© Acta Chemie Scandinavia 611997) 967-201
double carbonates in the system KCO,-CaCO, have
bbeen found in wood ash in partly burned fir, hemlock
and other trees in the western United States.*°
Fairchildite has also been identified in ash from the
combustion of biomass fuels.”
The aim of the present work is to determine the
structure of the double carbonate K,Ca,(CO,)s by
single-crystal X-ray diffraction, and to present an accur~
ate powder X-ray diffraction pattern that can serve as a
reliable identification tool for this phase.
Experimental
‘Synthesis. The growth of single erystals of K3Ca3(COs)s
of sufficient size for single-crystal X-ray diffraction
investigations cannot easily be carried out under normal
pressure conditions. On the one hand, the material
decomposes at higher temperatures before melting, and
on the other, solution growth may fail due to the poor
solubility of the compounds. A high-pressure technique
using a sealed container seemed to present a more
promising approach. As long as the piston-cylinder tech-
nique was available for the single-crystal growth experi-
‘ment, a minimum pressure of 10 kbar had to be chosen
‘The melting point of 810°C at 1 kbar for KCa3(CO,)s
shifts to a higher temperature at 10 kbar if the compound
remains a stable phase, Therefore a temperature of 950°C
at 10kbar was assumed to be still in the sub-solidus
region. A stoichiometric mixture of dried K3CO, (Baker)
and CaCO, (Riedel-deHatn) was prepared as a starting
material for K;Ca,(COs)s. This mixture was heated in
@ CO, atmosphere at 750°C for 11d. The new phase
was assumed to be pure since, according to an X-ray
387
WINBO ET AL
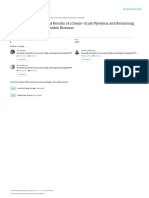
Fig. 1. A stereoscopic view of the structure per
(special postion a) and Ca(2) and Ki2) in gener
powder diffractogram, no traces of the starting compon-
ents or fairchildite and batschliite could be seen,
‘This material was then placed in a sealed gold capsule
that was heated at 950°C for 2h. The cooling was made
by slowly decreasing the temperature by 10°C! to
750°C. After reaching 750°C the experiment was
quenched simply by switching off the heater and releasing,
the pressure, The resulting crystals were checked with
powder X-ray diffraction, and were found to be identical
to the starting material
P-T calibrations of the piston-cylinder equipment were
‘made by DTA determinations of pressure temperature
effects on the melting point of LiCl. At any given pressure
the probable temperature uncertainty may be at most
48°C; at any temperature set point the pressure uncer-
tainty (always running the experiment in the piston-out
‘mode) may be considered to be +0.5 kbar.
X-Ray measurements. The material from the high-pres-
sure experiments could be described as pale yellow-green
granules, usually consisting of several intergrown crys-
tals. Preliminary investigations were performed on a
number of erystals, in the search for a suitable single
crystal. The impressions of intergrown crystals from
‘optical examination and of the twin lamellae reported
by Cooper er al were confirmed in Weissenberg and
precession photographs. A small fragment finaly turned
ut to be a single erystal suitable for an X-ray diffraction
investigation.
X-Ray intensity data were collected with a SYNTEX
3 automated four-cirele diffractometer using graphite-
monochromated MoK% radiation at 293K. The back-
ground was measured on each side of every reflection
for a time equal to the peak measurement. Cell para-
meters were determined by 25. reflections with
597-<20<21.92°. A psi-scan was carried out at 10
reflections selected from intensity measurement in the
range 5.94 < 26 < 52.20°, Experimental details are listed
in Table |
Calculations were cartied out using the Xtal3.2 pro-
ram package” OF a total of 8581 reflections measured,
816 unique reflections were used in the calculations. The
structure was solved using direct methods. Scattering
factors used (Ca?*, K*, O>, C®) were taken from
Ref, 10. A Lorenz polarisation correction and an empir-
ical absorption correction based on the psiscan were
388
;ndicula to the (001)-plane. Ca(1) and Kit) a
positions between the axes.
oxen
exu)
cain
oi
Yai
located on the three-fold axes
Table 1. Crystal and experimental data for KzCaz(COs)
Formula KeCag(COs)s
M, 338.38
Crystal system Trigonal
Space group 3 No. 148)
ah 13.0104)
86153)
1262:919)
6
267
Moka
0.71073
1025.62
Pale yellow-green
Crystal description
Crystal sizeimm 0.12 x 0.15 «0.08
Diffractometer: Nicolet R3 upgraded by
Siemens
Termporature/K 293
No, of refine for col
determination (20 range”)
25 (6.97 <20<21.92)
‘Sean mode 0-20
20 ranger” 5.96 <20< 602
hk range ~W
You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5810)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (844)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (348)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Latest Innovations in Pyrolysis TechnologyDocument15 pagesLatest Innovations in Pyrolysis TechnologypreemeeNo ratings yet
- PIP PNE00002 Piping Material Specification Selection, Development, and Application GuidelinesDocument17 pagesPIP PNE00002 Piping Material Specification Selection, Development, and Application Guidelinespreemee100% (3)
- 2000 - Removal of SOx and NOx Over Activated Carbon FibersDocument13 pages2000 - Removal of SOx and NOx Over Activated Carbon FiberspreemeeNo ratings yet
- 2014 - CO2 Capture Using Biochar Produced From Sugarcane Bagasse and Hickory WoodDocument27 pages2014 - CO2 Capture Using Biochar Produced From Sugarcane Bagasse and Hickory WoodpreemeeNo ratings yet
- 2020 - Ratio of H To C As Parameter To Predict Adsorption of Herbicides - LiuDocument41 pages2020 - Ratio of H To C As Parameter To Predict Adsorption of Herbicides - LiupreemeeNo ratings yet
- 2018 - Impact of Application of Biochar-Based Fertilizer On The Content of K and P in SoilDocument7 pages2018 - Impact of Application of Biochar-Based Fertilizer On The Content of K and P in SoilpreemeeNo ratings yet
- 2010 - Biochar in Agriculture - ICARDocument9 pages2010 - Biochar in Agriculture - ICARpreemeeNo ratings yet
- 2014 - Effects of Feedstock and Pyrolysis Temperature On Biochar Adsorption of Ammonium and NitrateDocument19 pages2014 - Effects of Feedstock and Pyrolysis Temperature On Biochar Adsorption of Ammonium and NitratepreemeeNo ratings yet
- Recent Advances in Biochar ApplicationDocument34 pagesRecent Advances in Biochar ApplicationpreemeeNo ratings yet
- 2008, Biochar in Northern Laos For Rice ProductionDocument4 pages2008, Biochar in Northern Laos For Rice ProductionpreemeeNo ratings yet
- 2020 - Use of Biochar in AgricultureDocument13 pages2020 - Use of Biochar in AgriculturepreemeeNo ratings yet
- Recent Perspectives in BiocharDocument19 pagesRecent Perspectives in BiocharpreemeeNo ratings yet
- Kemmer 10Document16 pagesKemmer 10preemeeNo ratings yet
- A Techincal Report On Design of Hot Oil System: June 2015Document25 pagesA Techincal Report On Design of Hot Oil System: June 2015preemeeNo ratings yet
- Biomass2Biochar Chapter11Document8 pagesBiomass2Biochar Chapter11preemeeNo ratings yet
- Biochar Production Through Slow Pyrolysis of Different Biomass Materials: Seeking The Best Operating ConditionsDocument31 pagesBiochar Production Through Slow Pyrolysis of Different Biomass Materials: Seeking The Best Operating ConditionspreemeeNo ratings yet
- Binder Neumann Paper 23rd 2015Document5 pagesBinder Neumann Paper 23rd 2015preemeeNo ratings yet
- ACS Energy Lett 2016 1 1149Document5 pagesACS Energy Lett 2016 1 1149preemeeNo ratings yet
- Amer Mineral 1974 59 353Document6 pagesAmer Mineral 1974 59 353preemeeNo ratings yet
- ACS Energy Lett 2017 2 454Document8 pagesACS Energy Lett 2017 2 454preemeeNo ratings yet
- PIP PNEM0001 - Piping Service Index PDFDocument5 pagesPIP PNEM0001 - Piping Service Index PDFpreemee100% (2)
- Amer Mineral 1973 58 778Document7 pagesAmer Mineral 1973 58 778preemeeNo ratings yet
- Acta Cryst 1961 14 1245 PDFDocument5 pagesActa Cryst 1961 14 1245 PDFpreemeeNo ratings yet