You might also like
- REGISTRATION FORM 7-A ManufacturerDocument5 pagesREGISTRATION FORM 7-A ManufacturerSuafa Traders100% (1)
- Iso853642009 (E)Document24 pagesIso853642009 (E)Suganth UNo ratings yet
- Wearable, Implantable, and Interventional Medical DevicesDocument18 pagesWearable, Implantable, and Interventional Medical DevicesMysha RahmanNo ratings yet
- RoadMap BBADocument4 pagesRoadMap BBARizwan AslamNo ratings yet
- EUDAMED MD Actor Module Q-A enDocument10 pagesEUDAMED MD Actor Module Q-A enGhada JlassiNo ratings yet
- Director Medical Devices R&D in NYC Resume Pallassana NarayananDocument7 pagesDirector Medical Devices R&D in NYC Resume Pallassana NarayananPallassanaNarayananNo ratings yet
- Marketing Mix Strategies for Medical DevicesDocument25 pagesMarketing Mix Strategies for Medical DevicesASHISH ANANDNo ratings yet
- REGULATORY SAMVADDocument19 pagesREGULATORY SAMVADvibhu yadavNo ratings yet
- Medical Devices Trendmapping: Opportunities in an Ageing EU MarketDocument7 pagesMedical Devices Trendmapping: Opportunities in an Ageing EU MarketEnrique Nieto CruzNo ratings yet
- Asia Language Medical Device Labeling RequirementsDocument2 pagesAsia Language Medical Device Labeling RequirementsJohn BabyNo ratings yet
- Enabling healthier future medical devicesDocument12 pagesEnabling healthier future medical devicesShrihariKMNo ratings yet
- Bulletin Healthcare Organisations 03142023Document46 pagesBulletin Healthcare Organisations 03142023dodyNo ratings yet
- QCI - AIMED Voluntary Initiative On Medical Devices: Technical Criteria For Certification of Medical Devices - ICMED 9000Document20 pagesQCI - AIMED Voluntary Initiative On Medical Devices: Technical Criteria For Certification of Medical Devices - ICMED 9000SureshNo ratings yet
- Medical Device RegulationsDocument88 pagesMedical Device Regulationssanthosh ezioNo ratings yet
- Imdrf Cons PPMDCDocument45 pagesImdrf Cons PPMDCAndres CañaveralNo ratings yet
- Medtronic MITG Investor Day PresentationDocument22 pagesMedtronic MITG Investor Day PresentationmedtechyNo ratings yet
- Industria 4.0 InglesDocument10 pagesIndustria 4.0 InglesFernando Gandolfo PérezNo ratings yet
- List of Red Medical Device ManufacturersDocument10 pagesList of Red Medical Device ManufacturersRuchik Shah0% (1)
- Class of Medical DevicesDocument14 pagesClass of Medical DevicesDuck Mann-ConsulNo ratings yet
- Medical Devices Rules India PDFDocument178 pagesMedical Devices Rules India PDFaman.4uNo ratings yet
- Human Factors in Medical DevicesDocument51 pagesHuman Factors in Medical Devicesajitbasrur445No ratings yet
- Medical Devices For The EU 070910Document38 pagesMedical Devices For The EU 070910Robert NatasorpNo ratings yet
- Unique Device Identification (UDI) System For Medical DevicesDocument14 pagesUnique Device Identification (UDI) System For Medical DevicesluNo ratings yet
- Medical Devices and Ehealth SolutionsDocument76 pagesMedical Devices and Ehealth SolutionsEliana Caceres TorricoNo ratings yet
- Medical Device Contract Manufacturing Global Market - Sample Report 25012022Document54 pagesMedical Device Contract Manufacturing Global Market - Sample Report 25012022Satish BirudukotaNo ratings yet
- ConsultantsDocument18 pagesConsultantsFredMelekzadaNo ratings yet
- COCIR Analysis On AI in Medical Device Legislation - Sept. 2020 - Final 2Document44 pagesCOCIR Analysis On AI in Medical Device Legislation - Sept. 2020 - Final 2Влад КасьяненкоNo ratings yet
- WEEE Regulations Guide for SMEsDocument33 pagesWEEE Regulations Guide for SMEsMahmoud YaseenNo ratings yet
- Cybersecurity of Medical DevicesDocument12 pagesCybersecurity of Medical DevicesppisupaNo ratings yet
- InterConnect2016 2824Document34 pagesInterConnect2016 2824Raja Churchill DassNo ratings yet
- The Indian Medical Device Industry - Nishith Desai AssociatesDocument66 pagesThe Indian Medical Device Industry - Nishith Desai AssociatesGulshan ChhibberNo ratings yet
- As en 540-2002 Clinical Investigation of Medical Devices For Human SubjectsDocument8 pagesAs en 540-2002 Clinical Investigation of Medical Devices For Human SubjectsSAI Global - APACNo ratings yet
- Amvex Intermittent Vacuum Regulator Manual PDFDocument4 pagesAmvex Intermittent Vacuum Regulator Manual PDFPeter ayalaNo ratings yet
- MDR Webinar 2016 PresentationDocument27 pagesMDR Webinar 2016 PresentationDavid MartinNo ratings yet
- Leitfaden Fuer App Entwickler enDocument65 pagesLeitfaden Fuer App Entwickler enloipoilNo ratings yet
- Echo Go HFDocument15 pagesEcho Go HFGaxi BofNo ratings yet
- Road MapDocument36 pagesRoad MapBernard Palarca Santos100% (1)
- Medtronic RTG Investor Day PresentationDocument36 pagesMedtronic RTG Investor Day PresentationmedtechyNo ratings yet
- Oven ManualDocument28 pagesOven ManualromwellNo ratings yet
- Vijay - Medical Device - System Engineer With 5 Plus YOEDocument6 pagesVijay - Medical Device - System Engineer With 5 Plus YOEVijay kumar BNo ratings yet
- Handbook - Classification of Medical DevicesDocument46 pagesHandbook - Classification of Medical DevicesRicha RohillaNo ratings yet
- Global Medical Device Nomenclature: Mark Wasmuth Secretary General GMDN AgencyDocument23 pagesGlobal Medical Device Nomenclature: Mark Wasmuth Secretary General GMDN AgencyluNo ratings yet
- SIEMENSDocument27 pagesSIEMENSKashish VyasNo ratings yet
- HPS Endoscope Decontamination 2004 PDFDocument190 pagesHPS Endoscope Decontamination 2004 PDFHong-Nam Kim100% (1)
- Vietnam medical device market poised for growthDocument11 pagesVietnam medical device market poised for growthÂn THiênNo ratings yet
- Indian Medical Device IndustryDocument12 pagesIndian Medical Device Industryraja manikumarNo ratings yet
- Exporting India 2011Document61 pagesExporting India 2011deepahireNo ratings yet
- Referance 2 Report PDFDocument7 pagesReferance 2 Report PDFMandhara KsNo ratings yet
- Medical Device Rules 2017 IndiaDocument108 pagesMedical Device Rules 2017 IndiaSubashiny Prabakaran0% (1)
- Biomedical TextileDocument19 pagesBiomedical TextileBetemariam limenewuNo ratings yet
- Case Study - VoicePicking: Henry Schein Distribution Centers Increase Efficiency Using Voice Picking TechnologyDocument2 pagesCase Study - VoicePicking: Henry Schein Distribution Centers Increase Efficiency Using Voice Picking TechnologyHenry ScheinNo ratings yet
- t09 - Software Verification and ValidationDocument64 pagest09 - Software Verification and ValidationophrqhbkxnuuhsmgcwNo ratings yet
- Human Reliability Assessment Ofr Medical DevicesDocument9 pagesHuman Reliability Assessment Ofr Medical DevicesGuilherme PagatiniNo ratings yet
- Production Linked Incentive (PLI) : A Well-Formulated SchemeDocument4 pagesProduction Linked Incentive (PLI) : A Well-Formulated SchemeSHIVAM SRIVASTAVANo ratings yet
- Medical Device IndustryDocument37 pagesMedical Device IndustrybasoormbNo ratings yet
- Fda Udi Unique Device Identifier GuidanceDocument11 pagesFda Udi Unique Device Identifier Guidanceqfbfabyhola100% (1)
- Find Doctors, Book Appointments with OlaDoc Digital Healthcare PlatformDocument2 pagesFind Doctors, Book Appointments with OlaDoc Digital Healthcare PlatformHassanNiazi100% (1)
- ISO-TC 210 - Quality Management and Corresponding General Aspects For Medical DevicesDocument17 pagesISO-TC 210 - Quality Management and Corresponding General Aspects For Medical DevicesIvanNo ratings yet
- Face Mask QuotationDocument2 pagesFace Mask QuotationSirNo ratings yet
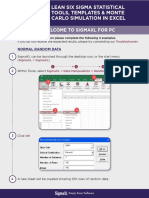
- Welcome To SigmaXL For PCDocument6 pagesWelcome To SigmaXL For PCNaveen KumarNo ratings yet
- Unlimited calls & 4G+ in EuropeDocument5 pagesUnlimited calls & 4G+ in EuropeNaveen KumarNo ratings yet
- MDSAP AS F0015.1.001 AO Nonconformity FlowchartDocument1 pageMDSAP AS F0015.1.001 AO Nonconformity FlowchartNaveen KumarNo ratings yet
- Sitem School Application FormDocument2 pagesSitem School Application FormNaveen KumarNo ratings yet
- Appreciating Systems Through an Organizational ViewDocument5 pagesAppreciating Systems Through an Organizational ViewNaveen KumarNo ratings yet
- Banana Pseudostem Value ChainDocument13 pagesBanana Pseudostem Value ChainNaveen Kumar0% (1)
- Translational Medicine and Biomedical Entrepreneurship ARTORG ScholarshipDocument4 pagesTranslational Medicine and Biomedical Entrepreneurship ARTORG ScholarshipNaveen KumarNo ratings yet
- BIC of CERN - Flyer - 2020Document4 pagesBIC of CERN - Flyer - 2020Naveen KumarNo ratings yet
- Professor/Mechanical Engineering, Rajalakshmi Engineering College, Thandalam, Chennai-602105. PH: 9790857137, 9940301665Document3 pagesProfessor/Mechanical Engineering, Rajalakshmi Engineering College, Thandalam, Chennai-602105. PH: 9790857137, 9940301665Naveen KumarNo ratings yet
- LogoDocument1 pageLogoNaveen KumarNo ratings yet
- This OneDocument3 pagesThis OneNaveen KumarNo ratings yet
- Me1401 NolDocument3 pagesMe1401 NolNaveen KumarNo ratings yet
- Ashok LeylandDocument8 pagesAshok LeylandNaveen KumarNo ratings yet
- App Format AFCAT MenDocument1 pageApp Format AFCAT MensodanforevaNo ratings yet
- TCVN 200-2011Document68 pagesTCVN 200-2011Vinh DaoNo ratings yet
- Air & Space SPDocument28 pagesAir & Space SPBharat JoshiNo ratings yet
- Tribune Delhi 20 Sep 2023Document14 pagesTribune Delhi 20 Sep 2023Vishal DhimanNo ratings yet
- 0051 001Document4 pages0051 001Raheem KassamNo ratings yet
- Topic 2: Burden of Proof: Common LawDocument11 pagesTopic 2: Burden of Proof: Common LawnicoleNo ratings yet
- Illustrator Cooperation AgreementDocument9 pagesIllustrator Cooperation AgreementCaroline FeijóNo ratings yet
- Snuggie TM Cancellation PleadingsDocument19 pagesSnuggie TM Cancellation PleadingsDaniel BallardNo ratings yet
- Auto Bus Transport Systems Inc V BautistaDocument2 pagesAuto Bus Transport Systems Inc V BautistaJordan Chavez100% (1)
- North Korea Through The Looking GlassDocument273 pagesNorth Korea Through The Looking GlassLiz T.100% (3)
- Certificate 1Document1 pageCertificate 1dnsent.123No ratings yet
- King Soopers Unfair Labor Practices FilingDocument3 pagesKing Soopers Unfair Labor Practices FilingMichael_Roberts2019No ratings yet
- Multiple Choice - Theory: Part 2Document4 pagesMultiple Choice - Theory: Part 2Nicolle JungNo ratings yet
- 2023 Term 2 - Marrung Koorie Community of PracticeDocument15 pages2023 Term 2 - Marrung Koorie Community of Practiceapi-337426672No ratings yet
- Creating PSLMC EO 180 s.1987Document2 pagesCreating PSLMC EO 180 s.1987Haniel GANo ratings yet
- 1 Veloso V DOLEDocument3 pages1 Veloso V DOLEDominique LarinNo ratings yet
- Jatiya Kabi Kazi Nazrul Islam University: Trishal, MymensinghDocument10 pagesJatiya Kabi Kazi Nazrul Islam University: Trishal, MymensinghrafatsantoNo ratings yet
- Law Enforcement Administration 1: Police Organization and Administration With Police PlanningDocument163 pagesLaw Enforcement Administration 1: Police Organization and Administration With Police PlanningJohn Leo R. Española,No ratings yet
- 509 Assignment-1Document11 pages509 Assignment-1mayani musumaliNo ratings yet
- Types of Budgets in Public AdministrationDocument4 pagesTypes of Budgets in Public AdministrationBluejoe SaidNo ratings yet
- Maths 2 Answer Sheet PDFDocument1 pageMaths 2 Answer Sheet PDFvinitha2675No ratings yet
- Case Study ON Maruti SuzukiDocument6 pagesCase Study ON Maruti SuzukianathomsanNo ratings yet
- Ian Ll. Macasinag Associates and M&M BLDG., First Park Subdivision, Daraga, Albay, PhilippinesDocument63 pagesIan Ll. Macasinag Associates and M&M BLDG., First Park Subdivision, Daraga, Albay, PhilippinesMaeJoyLoyolaBorlagdatanNo ratings yet
- Manila Jockey Club Employees Union V Manila Jockey ClubDocument2 pagesManila Jockey Club Employees Union V Manila Jockey ClubRejmali Good67% (3)
- Chap 1Document17 pagesChap 1akshat srivastavaNo ratings yet
- 4 Bill of ParticularsDocument2 pages4 Bill of ParticularsKetaks MooNo ratings yet
- Juvenile Delinquency in India Latest Trends and Entailing Amendments in Juvenile Justice ActpdfDocument19 pagesJuvenile Delinquency in India Latest Trends and Entailing Amendments in Juvenile Justice ActpdfMike WalterNo ratings yet
- IQ Level 3 Certificate For Working As A Close Protection Operative Within The Private Security IndustryDocument32 pagesIQ Level 3 Certificate For Working As A Close Protection Operative Within The Private Security IndustryBrolis LietuvisNo ratings yet
- Principle of Compensatory DiscriminationDocument2 pagesPrinciple of Compensatory DiscriminationsimranNo ratings yet
- Marcus Neiman Offers 'More Than The Conventional Approach' in Litigation - Daily Business ReviewDocument5 pagesMarcus Neiman Offers 'More Than The Conventional Approach' in Litigation - Daily Business ReviewMNRNo ratings yet
- Memorandum Circular No 78 1964Document18 pagesMemorandum Circular No 78 1964Sandy RoaNo ratings yet