You might also like
- Good Distribution Practices A Complete Guide - 2021 EditionFrom EverandGood Distribution Practices A Complete Guide - 2021 EditionNo ratings yet
- ISO 13485 Quality Management System A Complete Guide - 2020 EditionFrom EverandISO 13485 Quality Management System A Complete Guide - 2020 EditionNo ratings yet
- Validation Medical DeviceDocument69 pagesValidation Medical DeviceRakesh100% (6)
- Risk Management for Medical Device Manufacturers: [MD and IVD]From EverandRisk Management for Medical Device Manufacturers: [MD and IVD]No ratings yet
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionFrom EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNo ratings yet
- Comparison Chart of ISO 13485 and FDA QSR RequirementsDocument4 pagesComparison Chart of ISO 13485 and FDA QSR RequirementsjvivoloNo ratings yet
- Pharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersFrom EverandPharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersNo ratings yet
- Software Verification And Validation A Complete Guide - 2020 EditionFrom EverandSoftware Verification And Validation A Complete Guide - 2020 EditionNo ratings yet
- Medical Device Quality Management Systems: Strategy and Techniques for Improving Efficiency and EffectivenessFrom EverandMedical Device Quality Management Systems: Strategy and Techniques for Improving Efficiency and EffectivenessNo ratings yet
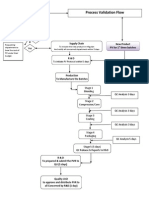
- Flow Chart Process ValidationDocument1 pageFlow Chart Process Validationmasthan6yNo ratings yet
- Sealing Process Validation Guideline - enDocument14 pagesSealing Process Validation Guideline - enyadu100% (1)
- MDSAP FAQ: Medical Device Single Audit Program Frequently Asked QuestionsDocument29 pagesMDSAP FAQ: Medical Device Single Audit Program Frequently Asked QuestionsloboufrjNo ratings yet
- Pharmaceutical Quality by Design: A Practical ApproachFrom EverandPharmaceutical Quality by Design: A Practical ApproachWalkiria S. SchlindweinNo ratings yet
- Software quality assurance Plan Complete Self-Assessment GuideFrom EverandSoftware quality assurance Plan Complete Self-Assessment GuideNo ratings yet
- ISO 9001 13485 and FDA QSR CompareDocument71 pagesISO 9001 13485 and FDA QSR CompareNoorm MENo ratings yet
- Medical Device Design: Innovation from Concept to MarketFrom EverandMedical Device Design: Innovation from Concept to MarketRating: 3 out of 5 stars3/5 (2)
- Equipment Qualification in the Pharmaceutical IndustryFrom EverandEquipment Qualification in the Pharmaceutical IndustryRating: 3.5 out of 5 stars3.5/5 (3)
- Production of Plasma Proteins for Therapeutic UseFrom EverandProduction of Plasma Proteins for Therapeutic UseRating: 3 out of 5 stars3/5 (5)
- Cleanroom Technology: Fundamentals of Design, Testing and OperationFrom EverandCleanroom Technology: Fundamentals of Design, Testing and OperationNo ratings yet
- Guide MDR GSPR RequirementsDocument38 pagesGuide MDR GSPR RequirementsMahmoud DomourNo ratings yet
- Establishing A CGMP Laboratory Audit System: A Practical GuideFrom EverandEstablishing A CGMP Laboratory Audit System: A Practical GuideNo ratings yet
- Process Validation For Medical Devices PDFDocument74 pagesProcess Validation For Medical Devices PDFPHÙ TRUNG TIÊN100% (4)
- Good Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsFrom EverandGood Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsNo ratings yet
- ISO 14971 A Complete Guide - 2021 EditionFrom EverandISO 14971 A Complete Guide - 2021 EditionRating: 1 out of 5 stars1/5 (1)
- Medical Device Design Validation SOPDocument1 pageMedical Device Design Validation SOPqmdocs0% (1)
- Audit Checklist For ISO 13485Document6 pagesAudit Checklist For ISO 13485EdNo ratings yet
- Comparison Matrix ISO 13485 To 21CFR820 R4Document64 pagesComparison Matrix ISO 13485 To 21CFR820 R4Tomasz Wojtera100% (1)
- SOP Equipment ValidationDocument15 pagesSOP Equipment Validationfarjana100% (7)
- QualityAgreementGuide2009 (Issued) FinalDocument24 pagesQualityAgreementGuide2009 (Issued) Finaldrs_mdu48100% (1)
- Cost-Contained Regulatory Compliance: For the Pharmaceutical, Biologics, and Medical Device IndustriesFrom EverandCost-Contained Regulatory Compliance: For the Pharmaceutical, Biologics, and Medical Device IndustriesNo ratings yet
- European MDR Readiness Checklist FillableDocument2 pagesEuropean MDR Readiness Checklist FillableHarold Braustein100% (2)
- TEMPLATE FOR PROCESS VALIDATION PROTOCOL - Pharmaceutical GuidanceDocument6 pagesTEMPLATE FOR PROCESS VALIDATION PROTOCOL - Pharmaceutical GuidancePackaging Development BernofarmNo ratings yet
- Tim Fields Master Validation PlanDocument7 pagesTim Fields Master Validation Planmanoj262400/2100% (1)
- Good Manufacturing Practices, Guidelines On ValidationDocument83 pagesGood Manufacturing Practices, Guidelines On ValidationPrince Moni100% (2)
- GMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsFrom EverandGMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsRating: 5 out of 5 stars5/5 (2)
- Operating Instructions Coding and Inspection Unit TQS-LM (Line Manager)Document94 pagesOperating Instructions Coding and Inspection Unit TQS-LM (Line Manager)BREWSKINo ratings yet
- Validating Cleaning Processes Used During The Manufacture of Medical DevicesDocument16 pagesValidating Cleaning Processes Used During The Manufacture of Medical DevicesFabricio Amorim100% (2)
- Ipf2018 Presentation21Document192 pagesIpf2018 Presentation21Vinoth JothiNo ratings yet
- Process Validation and Revalidation in Medical Device ProductionDocument7 pagesProcess Validation and Revalidation in Medical Device ProductionBREWSKINo ratings yet
- Validating Cleaning Processes Used During The Manufacture of Medical DevicesDocument16 pagesValidating Cleaning Processes Used During The Manufacture of Medical DevicesFabricio Amorim100% (2)
- Process Validation and Revalidation in Medical Device ProductionDocument7 pagesProcess Validation and Revalidation in Medical Device ProductionBREWSKINo ratings yet
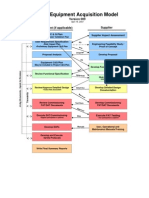
- AcquisitionModelVer9 PDFDocument5 pagesAcquisitionModelVer9 PDFantonygamalpharma100% (1)
- U R T Foraw R L F: Ser Equirements Emplate Ide Ange Iquid IllerDocument23 pagesU R T Foraw R L F: Ser Equirements Emplate Ide Ange Iquid IllerBREWSKINo ratings yet
- Process Validation and Revalidation in Medical Device ProductionDocument7 pagesProcess Validation and Revalidation in Medical Device ProductionBREWSKINo ratings yet
- URS Contents: Blank TemplateDocument11 pagesURS Contents: Blank TemplateBREWSKI100% (1)
- Urs Template ContentsDocument8 pagesUrs Template ContentsipatoffNo ratings yet
- Automatic Capper User RequirementsDocument26 pagesAutomatic Capper User RequirementsBREWSKINo ratings yet
- JETT URS Preparation Process GuideDocument7 pagesJETT URS Preparation Process GuideBREWSKINo ratings yet
- Risk MGT Template IT Pharma Validation EuropeDocument11 pagesRisk MGT Template IT Pharma Validation EuropeBREWSKINo ratings yet
- Urs Template ContentsDocument8 pagesUrs Template ContentsipatoffNo ratings yet
- NT - Akivision A CFRDocument60 pagesNT - Akivision A CFRBREWSKINo ratings yet
- Cleaning Validation StandardDocument14 pagesCleaning Validation StandardBREWSKINo ratings yet
- Risk MGT Template IT Pharma Validation EuropeDocument11 pagesRisk MGT Template IT Pharma Validation EuropeBREWSKINo ratings yet
- NT - Akivision A CFRDocument60 pagesNT - Akivision A CFRBREWSKINo ratings yet
- Sealing Process Validation Guideline - enDocument14 pagesSealing Process Validation Guideline - enyadu100% (1)
- JETT Document Status Tracking WorksheetDocument6 pagesJETT Document Status Tracking WorksheetBREWSKINo ratings yet
- Change Control Risk AssessmentDocument1 pageChange Control Risk AssessmentBREWSKINo ratings yet
- NT - Akivision A CFRDocument60 pagesNT - Akivision A CFRBREWSKINo ratings yet
- Change Control Risk AssessmentDocument1 pageChange Control Risk AssessmentBREWSKINo ratings yet
- CRBWhitepaperSerialization eDocument8 pagesCRBWhitepaperSerialization eBREWSKINo ratings yet
- CPV Case Study Interactive VersionDocument52 pagesCPV Case Study Interactive VersionBREWSKINo ratings yet
- Statistical Methods Overview for Pharmaceutical ProcessesDocument137 pagesStatistical Methods Overview for Pharmaceutical ProcessesDyah W K Antiek100% (1)
- Whitepaper Serialisation 150127Document3 pagesWhitepaper Serialisation 150127BREWSKINo ratings yet
- Texwipe PDA Cleaning and Cleaning Validation Chapter19Document26 pagesTexwipe PDA Cleaning and Cleaning Validation Chapter19davincicode888100% (1)
- Voip Ceiling SpeakerDocument42 pagesVoip Ceiling SpeakerMaría dieciochoNo ratings yet
- Vendors for H.T. Line MaterialsDocument587 pagesVendors for H.T. Line MaterialsAnonymous RjIueYSlNo ratings yet
- The Impact of Digital Advertising On Consumer Purchase DecisionsDocument11 pagesThe Impact of Digital Advertising On Consumer Purchase DecisionsyusufNo ratings yet
- Brand Rex LinkClickDocument170 pagesBrand Rex LinkClickniantonsNo ratings yet
- Review Paper On CFD Analysis of Electrical Vehicle Battery PackDocument4 pagesReview Paper On CFD Analysis of Electrical Vehicle Battery PackSikander Ahmed JahangirNo ratings yet
- Ar-One: Monitor Any Frequency From 10 KHZ To 3.3 GHZDocument2 pagesAr-One: Monitor Any Frequency From 10 KHZ To 3.3 GHZaliaj2No ratings yet
- AM612 PIR sensor manualDocument10 pagesAM612 PIR sensor manualEloy TorresNo ratings yet
- Omnivision III ASEBI-Service and Installation Guide-English-47753-0316 - CCv0003Document452 pagesOmnivision III ASEBI-Service and Installation Guide-English-47753-0316 - CCv0003Yerkin Martinez100% (1)
- Mba Ii Semester Assignment (Batch 2018-20) : SpecificationsDocument6 pagesMba Ii Semester Assignment (Batch 2018-20) : Specificationsakshara pradeepNo ratings yet
- Resume 07.11Document3 pagesResume 07.11KANNADIGA ANIL KERURKARNo ratings yet
- Assign02 - Solved Problems - Thin-Walled Pressure VesselsDocument12 pagesAssign02 - Solved Problems - Thin-Walled Pressure VesselsRAQUISIAGA, KYNAH YVE A.No ratings yet
- Nutanix - VMware Private Cloud Mods - ReqsDocument1 pageNutanix - VMware Private Cloud Mods - ReqsKeng Oei QuekNo ratings yet
- Istoria CMM (In Engleza) PDFDocument20 pagesIstoria CMM (In Engleza) PDF26110100% (1)
- Block Diagram: X541UV Repair GuideDocument7 pagesBlock Diagram: X541UV Repair GuideIlham PanjiNo ratings yet
- Chapter 16: Security: Silberschatz, Galvin and Gagne ©2018 Operating System Concepts - 10 EditionDocument58 pagesChapter 16: Security: Silberschatz, Galvin and Gagne ©2018 Operating System Concepts - 10 EditionJaswanth RachaNo ratings yet
- SMAD Review U1Document2 pagesSMAD Review U1David GhaliNo ratings yet
- Software Testing BooksDocument13 pagesSoftware Testing BooksNenad PetrövicNo ratings yet
- 24 Ways To Make Money With Chatgpt 1Document26 pages24 Ways To Make Money With Chatgpt 1PLANET CELLULLOID100% (1)
- Trane Pacu Voyager RT-PRC028AH-EN - 12152021Document156 pagesTrane Pacu Voyager RT-PRC028AH-EN - 12152021brian mmec2020No ratings yet
- Programmer Analyst As400 RPG SAP ABAPDocument3 pagesProgrammer Analyst As400 RPG SAP ABAPapi-121344987No ratings yet
- Wireless Security Design PDFDocument8 pagesWireless Security Design PDFIanRahmadiNo ratings yet
- Lean Six Sigma - WikipediaDocument17 pagesLean Six Sigma - WikipediaSAIBAL JANANo ratings yet
- Continuity/Insulation Resistance Test FormDocument1 pageContinuity/Insulation Resistance Test FormVictor BiacoloNo ratings yet
- J K Shah Classes Inter CA Nov 2022 TimetableDocument2 pagesJ K Shah Classes Inter CA Nov 2022 TimetableMegha ChauhanNo ratings yet
- 1ST Summative TestDocument4 pages1ST Summative TestFeo FranciscoNo ratings yet
- Machine Conditionvibration MonitoringDocument32 pagesMachine Conditionvibration Monitoringmohamed abourayaNo ratings yet
- BepDocument4 pagesBepkashifwarsiNo ratings yet
- MPLab Tutorial v1Document45 pagesMPLab Tutorial v1ASIM RIAZNo ratings yet
- E2e IT ProfileDocument4 pagesE2e IT ProfileDhiraj GautameNo ratings yet
- (Elec) Generator Excitation SystemDocument51 pages(Elec) Generator Excitation SystemThanh Son100% (1)










![Risk Management for Medical Device Manufacturers: [MD and IVD]](https://imgv2-1-f.scribdassets.com/img/word_document/602872428/149x198/825d3b5cd7/1666718194?v=1)