You might also like
- Renaissance RapierDocument66 pagesRenaissance Rapierrshpr100% (1)
- Wind Turbine Power Plant Seminar ReportDocument32 pagesWind Turbine Power Plant Seminar ReportShafieul mohammadNo ratings yet
- Optimal Control For Mathematical Models of Cancer Therapies - An Application of Geometric MethodsDocument511 pagesOptimal Control For Mathematical Models of Cancer Therapies - An Application of Geometric MethodskilgraveNo ratings yet
- Survival Analysis OverviewDocument23 pagesSurvival Analysis OverviewLoraline YuNo ratings yet
- Biostatistics LecturesDocument26 pagesBiostatistics LecturesSelin SakarNo ratings yet
- Am Jetstream Pre-Int Unit 8 Lesson 2Document2 pagesAm Jetstream Pre-Int Unit 8 Lesson 2Jennyfer Guevara50% (2)
- Trockel Flash Art 1987Document4 pagesTrockel Flash Art 1987cvg_geNo ratings yet
- Tata Steel LTD.: Elements Unit Min Max RemarksDocument2 pagesTata Steel LTD.: Elements Unit Min Max RemarksPavan KumarNo ratings yet
- Applicable Analysis: To Cite This Article: Zhaosheng Feng & Yubing Wan (2010) : Linearizing Transformations To ADocument18 pagesApplicable Analysis: To Cite This Article: Zhaosheng Feng & Yubing Wan (2010) : Linearizing Transformations To AalirafiqNo ratings yet
- Bajzer Z. Mathematical Modeling of Tumor Growth KineticsDocument45 pagesBajzer Z. Mathematical Modeling of Tumor Growth KineticsMario PuppiNo ratings yet
- Cancer Res 1988 Norton 7067 71Document6 pagesCancer Res 1988 Norton 7067 71Samuel Edhi Suranta SebayangNo ratings yet
- Cross-Sectional DependenceDocument5 pagesCross-Sectional DependenceIrfan UllahNo ratings yet
- Why Growth of Cancerous Tumors Is Gompertzian - A Symmetry-Based ExplanationDocument9 pagesWhy Growth of Cancerous Tumors Is Gompertzian - A Symmetry-Based ExplanationFrancis ZhouNo ratings yet
- Emation of Survival Based On Proportional Hazards When Cure Is A Possibility-A - TSODIKOV - 2001Document10 pagesEmation of Survival Based On Proportional Hazards When Cure Is A Possibility-A - TSODIKOV - 2001Ali HamliliNo ratings yet
- NIH Public Access: Adaptive TherapyDocument22 pagesNIH Public Access: Adaptive Therapybdvd1007092No ratings yet
- Quantec 02Document7 pagesQuantec 02onco100% (1)
- Optimal Mutation Rates and Selection Pressure in Genetic AlgorithmsDocument8 pagesOptimal Mutation Rates and Selection Pressure in Genetic AlgorithmsArganta SamudraNo ratings yet
- Statistical Treatment of Data, Data Interpretation, and ReliabilityDocument6 pagesStatistical Treatment of Data, Data Interpretation, and Reliabilitydraindrop8606No ratings yet
- Herschlag RsDocument6 pagesHerschlag RsValdir JúniorNo ratings yet
- Ghaffari 2016Document15 pagesGhaffari 2016TomyNo ratings yet
- On The Predictability of Infectious Disease Outbreaks: ArticleDocument8 pagesOn The Predictability of Infectious Disease Outbreaks: ArticleAlessandra PelliniNo ratings yet
- CMMM2014 172923Document12 pagesCMMM2014 172923nilufarkomiljonovna21No ratings yet
- Applications of Mathematics To Biology: Manuscript ID: #0443 Original Research PaperDocument5 pagesApplications of Mathematics To Biology: Manuscript ID: #0443 Original Research Paperrizka arzitaNo ratings yet
- Kidney TumourDocument5 pagesKidney Tumourlogu_thalirNo ratings yet
- Estimation of Simultaneous Systems of Spatially Interrelated Cross Sectional EquationsDocument24 pagesEstimation of Simultaneous Systems of Spatially Interrelated Cross Sectional Equationsjuan carlos molano toroNo ratings yet
- Royal Stata Society Series C - 2022 - Eletti - A Unifying Framework For Flexible Excess Hazard Modelling With ApplicationsDocument19 pagesRoyal Stata Society Series C - 2022 - Eletti - A Unifying Framework For Flexible Excess Hazard Modelling With ApplicationsJuan Fernando SubiranaNo ratings yet
- Stochastic Model For Cancer Cell Growth With Spontaneous Mutation and ProliferationDocument11 pagesStochastic Model For Cancer Cell Growth With Spontaneous Mutation and ProliferationLekshmiNo ratings yet
- Cancer - November December 1961 - Schwartz - A Biomathematical Approach To Clinical Tumor GrowthDocument23 pagesCancer - November December 1961 - Schwartz - A Biomathematical Approach To Clinical Tumor Growthranisingh4760No ratings yet
- Transactions of Society of Actuaries 1 9 9 5 VOL. 47Document26 pagesTransactions of Society of Actuaries 1 9 9 5 VOL. 47Angel Tec LópezNo ratings yet
- 10.3934 Mbe.2017004Document22 pages10.3934 Mbe.2017004zulfaaakfiiNo ratings yet
- Articulo 2.Document17 pagesArticulo 2.WILMAR SEBASTIAN FLECHAS SUAREZNo ratings yet
- Cure Rate ModelDocument10 pagesCure Rate Modelcarlton smithNo ratings yet
- Mathematical Modeling of Cancer RadiovirotherapyDocument24 pagesMathematical Modeling of Cancer Radiovirotherapylevi trianaNo ratings yet
- Theoretical Biology and Medical ModellingDocument6 pagesTheoretical Biology and Medical ModellingManjunath RamachandraNo ratings yet
- Johansen Cointegration Test MethodDocument19 pagesJohansen Cointegration Test MethodIhtisham JadoonNo ratings yet
- Bayesian Meta-Analysis For Longitudinal Models Using Multivariate Mixture PriorsDocument10 pagesBayesian Meta-Analysis For Longitudinal Models Using Multivariate Mixture Priorstem o0pNo ratings yet
- Krijkamp Et Al. - 2020 - A Multidimensional Array Representation of State-Transition Model DynamicsDocument7 pagesKrijkamp Et Al. - 2020 - A Multidimensional Array Representation of State-Transition Model DynamicsferalaesNo ratings yet
- Entropy: Quantum and Ecosystem EntropiesDocument13 pagesEntropy: Quantum and Ecosystem EntropiesjohnnirbanoNo ratings yet
- Accelerated Tumor Invasion Under Non-Isotropic Cell Dispersal in GlioblastomasDocument11 pagesAccelerated Tumor Invasion Under Non-Isotropic Cell Dispersal in GlioblastomasDemian PereiraNo ratings yet
- DeepSurv Using A Cox Proportional Hasards DeepNets 1652051740Document12 pagesDeepSurv Using A Cox Proportional Hasards DeepNets 1652051740nwillknow7No ratings yet
- Journal of Biomechanics: W. Sun, J.H. Jin, M.P. Reed, F.S. Gayzik, K.A. Danelson, C.R. Bass, J.Y. Zhang, J.D. RuppDocument8 pagesJournal of Biomechanics: W. Sun, J.H. Jin, M.P. Reed, F.S. Gayzik, K.A. Danelson, C.R. Bass, J.Y. Zhang, J.D. RuppStefanita CiunelNo ratings yet
- Optimal Control Analysis of A Mathematical Model For Breast CancerDocument28 pagesOptimal Control Analysis of A Mathematical Model For Breast CancerranaNo ratings yet
- Rockne Et Al - Brain TumorDocument18 pagesRockne Et Al - Brain TumorDaniela SanturioNo ratings yet
- Metodos AnaliticosDocument136 pagesMetodos AnaliticosCarlos Velando CabañasNo ratings yet
- Pred Prey Tumor ModelDocument8 pagesPred Prey Tumor Modelddddd34r54tdNo ratings yet
- The Top-K Tau-Path Screen For Monotone Association: Yu Et Al. 2011Document40 pagesThe Top-K Tau-Path Screen For Monotone Association: Yu Et Al. 2011Yusuf RihabeNo ratings yet
- Cointegracion MacKinnon, - Et - Al - 1996 PDFDocument26 pagesCointegracion MacKinnon, - Et - Al - 1996 PDFAndy HernandezNo ratings yet
- Estimating Tumor Growth Rates in VivoDocument27 pagesEstimating Tumor Growth Rates in VivoSamuel Edhi Suranta SebayangNo ratings yet
- Modeling and Simulating Chemical Reactions: 1 MotivationDocument31 pagesModeling and Simulating Chemical Reactions: 1 MotivationakumcNo ratings yet
- A New Method To Study Stochastic Growth Equations: Application To The Edwards-Wilkinson EquationDocument4 pagesA New Method To Study Stochastic Growth Equations: Application To The Edwards-Wilkinson Equationjuanjo romeroNo ratings yet
- Literature Review On Canonical Correlation AnalysisDocument8 pagesLiterature Review On Canonical Correlation Analysisfveec9sxNo ratings yet
- Feller 1949 - On The Theory of Stochastic Processes, With Par - Ticular Reference To ApplicationsDocument30 pagesFeller 1949 - On The Theory of Stochastic Processes, With Par - Ticular Reference To ApplicationsGugo ManNo ratings yet
- Mathematical CancerDocument14 pagesMathematical CancerTerapi Suara IndonesiaNo ratings yet
- MainDocument16 pagesMainFeby ArdhaniNo ratings yet
- Science 2012 Sugihara 496 500 PDFDocument5 pagesScience 2012 Sugihara 496 500 PDFErnesto Lara HernándezNo ratings yet
- Some Insight On Censored Cost Estimators: H. Zhao, Y. Cheng and H. BangDocument8 pagesSome Insight On Censored Cost Estimators: H. Zhao, Y. Cheng and H. Bangalx_666666No ratings yet
- Dynamics of Tumor Interaction With The Host Immune SystemDocument16 pagesDynamics of Tumor Interaction With The Host Immune SystemSyed Tahseen RazaNo ratings yet
- Limits To Causal Inference With State-Space Reconstruction For Infectious DiseaseDocument32 pagesLimits To Causal Inference With State-Space Reconstruction For Infectious DiseaseCodeLux theGr8No ratings yet
- Pnas 2007 Gerrish 6266 71Document6 pagesPnas 2007 Gerrish 6266 71api-211640838No ratings yet
- An Introduction To Viability Theory and Management of Renewable ResourcesDocument42 pagesAn Introduction To Viability Theory and Management of Renewable ResourcesAmine SlavousNo ratings yet
- Alvin M. Saperstein, The Prediction of Unpredictability: Applications of The New Paradigm of Chaos in Dynamical Systems To The Old Problem of The Stability of A System of Hostile NationsDocument25 pagesAlvin M. Saperstein, The Prediction of Unpredictability: Applications of The New Paradigm of Chaos in Dynamical Systems To The Old Problem of The Stability of A System of Hostile NationsAnonymous hXQMt5No ratings yet
- Review of Fundamental Problems and Solutions in Fractal Surface GrowthDocument26 pagesReview of Fundamental Problems and Solutions in Fractal Surface GrowthChristian LauNo ratings yet
- Disease Transmission Dynamics of An Epidemiological Predator-Prey System in Open Advective EnvironmentsDocument23 pagesDisease Transmission Dynamics of An Epidemiological Predator-Prey System in Open Advective EnvironmentsAlex FernandezNo ratings yet
- 2023 07 04 547696v1 FullDocument21 pages2023 07 04 547696v1 FullasdfsoidjfoiqjwefNo ratings yet
- Optimal Control of Joint Multi-Virus Infection and Information SpreadingDocument6 pagesOptimal Control of Joint Multi-Virus Infection and Information Spreadinglevi trianaNo ratings yet
- Operations Research For Health Care: Abdulfatai A. Momoh, Armin FügenschuhDocument13 pagesOperations Research For Health Care: Abdulfatai A. Momoh, Armin Fügenschuhlevi trianaNo ratings yet
- Avascular Growth, Angiogenesis and Vascular Growth in Solid Tumours: The Mathematical Modelling of The Stages of Tumour DevelopmentDocument41 pagesAvascular Growth, Angiogenesis and Vascular Growth in Solid Tumours: The Mathematical Modelling of The Stages of Tumour Developmentlevi trianaNo ratings yet
- One Health: SciencedirectDocument6 pagesOne Health: Sciencedirectlevi trianaNo ratings yet
- Mathematical Modeling of Cancer RadiovirotherapyDocument24 pagesMathematical Modeling of Cancer Radiovirotherapylevi trianaNo ratings yet
- 1 s2.0 S0887233319305661 MainDocument9 pages1 s2.0 S0887233319305661 Mainlevi trianaNo ratings yet
- Informatics in Medicine Unlocked: Joseph Malinzi, Kevin Bosire Basita, Sara Padidar, Henry Ademola AdeolaDocument15 pagesInformatics in Medicine Unlocked: Joseph Malinzi, Kevin Bosire Basita, Sara Padidar, Henry Ademola Adeolalevi trianaNo ratings yet
- Classification by Depth Distribution of Phytoplankton and ZooplanktonDocument31 pagesClassification by Depth Distribution of Phytoplankton and ZooplanktonKeanu Denzel BolitoNo ratings yet
- CanagliflozinDocument7 pagesCanagliflozin13201940No ratings yet
- Chemistry Bridging Course Lecture NotesDocument3 pagesChemistry Bridging Course Lecture NotesNNo ratings yet
- Inspection and Test Plan (ITP) For Spherical Storage Tanks: Dehloran Olefin PlantDocument9 pagesInspection and Test Plan (ITP) For Spherical Storage Tanks: Dehloran Olefin PlantbahmanNo ratings yet
- Aeroacoustic Optimization of Wind Turbine Airfoils by Combining Thermographic and Acoustic Measurement DataDocument4 pagesAeroacoustic Optimization of Wind Turbine Airfoils by Combining Thermographic and Acoustic Measurement DatamoussaouiNo ratings yet
- Sociology Internal AssessmentDocument21 pagesSociology Internal AssessmentjavoughnNo ratings yet
- Safelisting in Office 365Document5 pagesSafelisting in Office 365Brett ThomasNo ratings yet
- Gopesh Obalappa Pilot ResumeDocument3 pagesGopesh Obalappa Pilot ResumeGopesh ObalappaNo ratings yet
- If Else ExercisesDocument5 pagesIf Else ExercisesHoney Jean PerezNo ratings yet
- Department of Labor: BC Bond ListDocument67 pagesDepartment of Labor: BC Bond ListUSA_DepartmentOfLabor100% (1)
- ECON 211: Principles of Macroeconomics-901: Smhussain@vcu - EduDocument6 pagesECON 211: Principles of Macroeconomics-901: Smhussain@vcu - EdusshinnNo ratings yet
- 2006 317Document13 pages2006 317LonginoNo ratings yet
- 78EZDM Product Specifications Conector para Cable de 7 OctavosDocument4 pages78EZDM Product Specifications Conector para Cable de 7 OctavosEdwin Santiago QuispeNo ratings yet
- Group 6G Revised Research Manuscript 1Document57 pagesGroup 6G Revised Research Manuscript 1Mc Rollyn VallespinNo ratings yet
- Inertia Physics: Defi Ni Ti OnDocument2 pagesInertia Physics: Defi Ni Ti OnSentoash NaiduNo ratings yet
- Mutant ChroniclesDocument3 pagesMutant ChroniclesZoth BernsteinNo ratings yet
- Cassius Resume CVLatestDocument3 pagesCassius Resume CVLatestCaszNo ratings yet
- VW 60330 2009 12 eDocument29 pagesVW 60330 2009 12 eAmir Borhanipour100% (1)
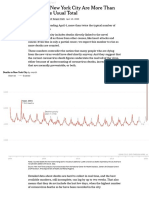
- Deaths in New York City Are More Than Double The Usual TotalDocument3 pagesDeaths in New York City Are More Than Double The Usual TotalRamón RuizNo ratings yet
- Prakhar Gupta Epics and Empires-Game of Thrones Make Up EssayDocument5 pagesPrakhar Gupta Epics and Empires-Game of Thrones Make Up EssayGat DanNo ratings yet
- Wooden Buildings: exposed to tiếp xúc với dramatic renewal sự làm mới đáng kểDocument6 pagesWooden Buildings: exposed to tiếp xúc với dramatic renewal sự làm mới đáng kểNguyễn Phạm Thảo NguyênNo ratings yet
- 3286306Document5 pages3286306Sanjit OracleTrainingNo ratings yet
- Kinetika Kimia Pada Laju ReaksiDocument25 pagesKinetika Kimia Pada Laju ReaksiWardahNo ratings yet
- Naaladiyar: Watch Free MoviesDocument6 pagesNaaladiyar: Watch Free MoviesVijayaLakshmi IyerNo ratings yet
- Generation Transmission and Distribution Notes.Document88 pagesGeneration Transmission and Distribution Notes.Abhishek NirmalkarNo ratings yet