You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Iso 16948-2015 固体生物燃料的碳、氢和氮的测定法Document16 pagesIso 16948-2015 固体生物燃料的碳、氢和氮的测定法Winnjone YinNo ratings yet
- 104.GB 8076-2008Document27 pages104.GB 8076-2008Winnjone YinNo ratings yet
- DLT 852-2016 enDocument75 pagesDLT 852-2016 enWinnjone YinNo ratings yet
- Astm d1857 (m) -2010 Aft 灰熔融Document5 pagesAstm d1857 (m) -2010 Aft 灰熔融Winnjone YinNo ratings yet
- ISO 540-2008 Hard Coal and Coke Determination of Ash Fusibility 灰熔点Document18 pagesISO 540-2008 Hard Coal and Coke Determination of Ash Fusibility 灰熔点Winnjone YinNo ratings yet
- Iso 11722 通氮测试煤中水分Document10 pagesIso 11722 通氮测试煤中水分Winnjone YinNo ratings yet
- Iso 589-2008 煤的总水分测定Document16 pagesIso 589-2008 煤的总水分测定Winnjone YinNo ratings yet
- ISO 29541-2010 Solid Mineral Fuels - Determination Total Carbon Hydrogen NitrogDocument18 pagesISO 29541-2010 Solid Mineral Fuels - Determination Total Carbon Hydrogen NitrogWinnjone YinNo ratings yet
- ASTM D346 (M) −2011 Collection and Preparation of Coke Samples for Laboratory Analysis 焦炭的采集与准备Document5 pagesASTM D346 (M) −2011 Collection and Preparation of Coke Samples for Laboratory Analysis 焦炭的采集与准备Winnjone YinNo ratings yet
- Iso 5068-2-2007Document12 pagesIso 5068-2-2007Winnjone YinNo ratings yet
- Iso 5068-1-2007Document14 pagesIso 5068-1-2007Winnjone YinNo ratings yet
- ISO 5071 1 2013 褐煤挥发份Document19 pagesISO 5071 1 2013 褐煤挥发份Winnjone YinNo ratings yet
- ASTM D197−87 (2012) Sampling and Fineness Test of Pulverized Coal 取样和粒度分析Document7 pagesASTM D197−87 (2012) Sampling and Fineness Test of Pulverized Coal 取样和粒度分析Winnjone YinNo ratings yet
- ISO 1171-2010 煤中灰分Document12 pagesISO 1171-2010 煤中灰分Winnjone YinNo ratings yet
- Astm D3175-2011Document6 pagesAstm D3175-2011Winnjone YinNo ratings yet
- ASTM D5865 2013 Gross Calorific Value of Coal and CokeDocument19 pagesASTM D5865 2013 Gross Calorific Value of Coal and CokeWinnjone YinNo ratings yet
- Astm d5865 2012Document19 pagesAstm d5865 2012Winnjone YinNo ratings yet
- ASTM D3302 M - 2012 Total MoistureDocument8 pagesASTM D3302 M - 2012 Total MoistureWinnjone YinNo ratings yet
- ASTM D3172−13 Proximate Analysis of Coal and Coke 煤和焦炭的近似分析Document2 pagesASTM D3172−13 Proximate Analysis of Coal and Coke 煤和焦炭的近似分析Winnjone YinNo ratings yet
- Astm D3174 2012Document6 pagesAstm D3174 2012Winnjone YinNo ratings yet
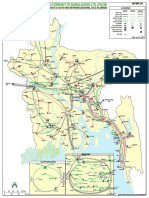
- 孟加拉电网分布图 PDFDocument1 page孟加拉电网分布图 PDFWinnjone YinNo ratings yet
- Astm D3173 2011Document4 pagesAstm D3173 2011Winnjone YinNo ratings yet
- 孟加拉电网分布图 PDFDocument1 page孟加拉电网分布图 PDFWinnjone YinNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Orgel DiagramsDocument1 pageOrgel Diagramsmahi kumarNo ratings yet
- Polymer Notes 2017Document10 pagesPolymer Notes 2017Bikram YadavNo ratings yet
- APR1400 SSAR SupplementDocument199 pagesAPR1400 SSAR SupplementAbdalrahman Taha (Jíɾaïya)No ratings yet
- Tank Breather Filter With Filler Strainer ELF: Up To 5500 L/minDocument6 pagesTank Breather Filter With Filler Strainer ELF: Up To 5500 L/minThanh CongNo ratings yet
- Physicochemical and Functional Properties of Soy Protein Isolate As A Function of Water Activity and StorageDocument9 pagesPhysicochemical and Functional Properties of Soy Protein Isolate As A Function of Water Activity and StoragePaul Jefferson Flores HurtadoNo ratings yet
- Autothermal Reforming of Methane Design and PerforDocument7 pagesAutothermal Reforming of Methane Design and Perforromi moriNo ratings yet
- Section: Sopro Product Systems For Sustainable ConstructionDocument38 pagesSection: Sopro Product Systems For Sustainable ConstructionpankajNo ratings yet
- 907 1TDSDocument2 pages907 1TDSJainam Shah100% (1)
- Viscosity of Gases: Marcia L. Huber and Allan H. HarveyDocument2 pagesViscosity of Gases: Marcia L. Huber and Allan H. HarveyUmarNo ratings yet
- 5989-9667EN Splitler 7890BDocument8 pages5989-9667EN Splitler 7890BNatalia CruzNo ratings yet
- Practice Questions Topic 1.4 IB BiologyDocument12 pagesPractice Questions Topic 1.4 IB BiologySamantha DAIRONo ratings yet
- Dna Extraction Lab - by Alija KankamDocument13 pagesDna Extraction Lab - by Alija Kankamapi-457025605No ratings yet
- Tutorial 1 Chm256Document2 pagesTutorial 1 Chm256ANIS AMIRAH SALIMINNo ratings yet
- Understanding The Engineering Polymers. (Annealing of Polymers and Composites), Part 3Document3 pagesUnderstanding The Engineering Polymers. (Annealing of Polymers and Composites), Part 3juan k RestrepoNo ratings yet
- 1.2.4 HDG - Painting Issues PDFDocument2 pages1.2.4 HDG - Painting Issues PDFAnonymous 1AAjd0No ratings yet
- Pile Cap of Abutment A1 & A2 (Takarma) - 1Document1 pagePile Cap of Abutment A1 & A2 (Takarma) - 1Nilay GandhiNo ratings yet
- Qualitative Tests For CarbohydratesDocument2 pagesQualitative Tests For CarbohydratesChristopher GalivoNo ratings yet
- Welding Neseahch: Porosity and Hydrogen Absorption in Aluminum WeldsDocument12 pagesWelding Neseahch: Porosity and Hydrogen Absorption in Aluminum WeldsdietersimaNo ratings yet
- IC - Fab - Quiz 1Document5 pagesIC - Fab - Quiz 1Debdeep BhattacharyaNo ratings yet
- Preparation of Extractive-Free Wood: Standard Test Method ForDocument2 pagesPreparation of Extractive-Free Wood: Standard Test Method ForCasey RybackNo ratings yet
- Chem Q2 WRITTEN WORK 1Document6 pagesChem Q2 WRITTEN WORK 1leyt kanaNo ratings yet
- Chemistry 251 Lab NotesDocument8 pagesChemistry 251 Lab NotesAnupa GhoseNo ratings yet
- Sri Chaitanya IIT Academy., India.: Key SheetDocument38 pagesSri Chaitanya IIT Academy., India.: Key SheetAjay BhatnagarNo ratings yet
- CicaLiquid NH3 - L-020EDocument4 pagesCicaLiquid NH3 - L-020Eandr sonNo ratings yet
- TM 2 Fundamentals of Organic Chemistry 7th Edition by John McMurryDocument15 pagesTM 2 Fundamentals of Organic Chemistry 7th Edition by John McMurrysukma AsaNo ratings yet
- Icp-Ms 05022018Document37 pagesIcp-Ms 05022018pankajNo ratings yet
- Updated Client List J4690, 47,48 OnwardsDocument18 pagesUpdated Client List J4690, 47,48 Onwardshbgroups3018No ratings yet
- Classification of Different Welding Processes With PDFDocument4 pagesClassification of Different Welding Processes With PDFMadhu NNo ratings yet
- LA-159159-50 CidezymeDocument31 pagesLA-159159-50 CidezymeMarcerceNo ratings yet
- IB CHEMISTRY 1ed TR Worksheet AnsDocument47 pagesIB CHEMISTRY 1ed TR Worksheet AnsKelvin ChoyNo ratings yet