You might also like
- Alastair I. M. Rae Jim Napolitano Quantum Mechanics CRC Press 2015Document435 pagesAlastair I. M. Rae Jim Napolitano Quantum Mechanics CRC Press 2015Roman Sokolov100% (1)
- Transformation de LaplaceDocument20 pagesTransformation de LaplaceA.BenhariNo ratings yet
- Sucheta Mahajan - Independence and Partition - The Erosion of Colonial Power in India-SAGE Publications (2000)Document427 pagesSucheta Mahajan - Independence and Partition - The Erosion of Colonial Power in India-SAGE Publications (2000)Bitan kumar DeNo ratings yet
- Pykhtin MFADocument6 pagesPykhtin MFAhsch345No ratings yet
- Quantum Mechanics II - Homework 3Document7 pagesQuantum Mechanics II - Homework 3Ale GomezNo ratings yet
- Chap 6 SolutionsDocument34 pagesChap 6 Solutionsanonyme3897No ratings yet
- Lecture 5: The Non-Equilibrium Green Function MethodDocument27 pagesLecture 5: The Non-Equilibrium Green Function Methodslag12309No ratings yet
- 10 Greens FunctionDocument9 pages10 Greens FunctionAnonymous j6r5KRtrH2No ratings yet
- Dyson SeriesDocument25 pagesDyson SeriesNeymar MessiNo ratings yet
- 10 Greens FunctionDocument8 pages10 Greens FunctionDiego SilvaNo ratings yet
- EEE 303 HW # 1 SolutionsDocument22 pagesEEE 303 HW # 1 SolutionsDhirendra Kumar SinghNo ratings yet
- Metodo de HeunDocument6 pagesMetodo de HeunNilton Isaias Mariños HerreraNo ratings yet
- Second Set of Slides Notes PDFDocument23 pagesSecond Set of Slides Notes PDFJustina MweendiNo ratings yet
- Lecture14 Probset1.8Document4 pagesLecture14 Probset1.8JRNo ratings yet
- A Comparison Between Active and Passive Vibration Control of Non-Linear Simple Pendulum PDFDocument12 pagesA Comparison Between Active and Passive Vibration Control of Non-Linear Simple Pendulum PDFmohamed mourad LafifiNo ratings yet
- Interacting Quantum Fields C6, HT 2015: 1.1 Toy ModelsDocument25 pagesInteracting Quantum Fields C6, HT 2015: 1.1 Toy Modelscifarha venantNo ratings yet
- The Convolution Theorem: PrerequisitesDocument16 pagesThe Convolution Theorem: PrerequisitesFegason FegyNo ratings yet
- Template PendulumDocument7 pagesTemplate Pendulumjumana basselNo ratings yet
- One Dimension Free Fermions Green's Function: Ext ExtDocument6 pagesOne Dimension Free Fermions Green's Function: Ext ExtMatheus SchosslerNo ratings yet
- A New Method To Estimate A First-Order Plus Time Delay Model From Step ResponseDocument11 pagesA New Method To Estimate A First-Order Plus Time Delay Model From Step ResponseGetahun EsNo ratings yet
- Homotopy Perturbation Method For Time-FractionalDocument10 pagesHomotopy Perturbation Method For Time-Fractionalsingh_mathitbhu5790No ratings yet
- 4th SlidesDocument116 pages4th SlidesJonas XoxaeNo ratings yet
- 1 s2.0 S0377042707005nDocument18 pages1 s2.0 S0377042707005n青山漫步No ratings yet
- Notes LT2Document20 pagesNotes LT2Zaw Phyo OoNo ratings yet
- Dirac Comb and Flavors of Fourier Transforms: 1 Exp Ik2Document7 pagesDirac Comb and Flavors of Fourier Transforms: 1 Exp Ik2Lường Văn LâmNo ratings yet
- ProvaDocument4 pagesProvajog1No ratings yet
- The Time Evolution Operator As A Time-Ordered ExponentialDocument8 pagesThe Time Evolution Operator As A Time-Ordered ExponentialMonky PandaNo ratings yet
- Maximum Smoothness Forward Rate CurvesDocument25 pagesMaximum Smoothness Forward Rate CurvesbobmezzNo ratings yet
- Convolution 18.031, Haynes Miller and Jeremy OrloffDocument10 pagesConvolution 18.031, Haynes Miller and Jeremy OrloffPrineezyNo ratings yet
- Damped Simple Harmonic Motion - Critical DampingDocument13 pagesDamped Simple Harmonic Motion - Critical DampingRohan MohataNo ratings yet
- Convolution PDFDocument5 pagesConvolution PDFRommel AnacanNo ratings yet
- Numerical Solution of Damped Forced Oscillator Problem Using Haar WaveletsDocument12 pagesNumerical Solution of Damped Forced Oscillator Problem Using Haar WaveletsAbel NetoNo ratings yet
- Application 8Document4 pagesApplication 8Dr. Ir. R. Didin Kusdian, MT.No ratings yet
- Cal3 WS1 Space CurvesDocument6 pagesCal3 WS1 Space Curves陳泓睿No ratings yet
- Entropy: Quantum Dynamics in A Fluctuating EnvironmentDocument13 pagesEntropy: Quantum Dynamics in A Fluctuating EnvironmentkenfackNo ratings yet
- Notes On 1 Particle ScatteringDocument5 pagesNotes On 1 Particle ScatteringJuliana HarmatiukNo ratings yet
- Lec 0813Document10 pagesLec 0813Chevy TeoNo ratings yet
- Shehu TransformDocument10 pagesShehu TransformFaisal Jabbar Faisal JabbarNo ratings yet
- Fluctuation-Dissipation TheoremDocument2 pagesFluctuation-Dissipation TheoremGunnar CalvertNo ratings yet
- ME311 Autumn2021 T4Document2 pagesME311 Autumn2021 T4Varun PathakNo ratings yet
- The Finite Difference Method For Transient Convection-Diffusion ProblemsDocument10 pagesThe Finite Difference Method For Transient Convection-Diffusion ProblemsSamuelHericNo ratings yet
- 20 5 Convolution THMDocument8 pages20 5 Convolution THMsanjayNo ratings yet
- Data-Based Approach To Feedback-Feedforward Controller Design From Closed-Loop Plant DataDocument6 pagesData-Based Approach To Feedback-Feedforward Controller Design From Closed-Loop Plant DataArif HidayatNo ratings yet
- Taller Cuatro Análisis de SistemasDocument5 pagesTaller Cuatro Análisis de SistemasCarlosNo ratings yet
- Assignment 1 Communication Theory EE304: Submit QTS.: 4, 7, 10 (A, B, E), 12, 14, 16 and 17Document6 pagesAssignment 1 Communication Theory EE304: Submit QTS.: 4, 7, 10 (A, B, E), 12, 14, 16 and 17ayuNo ratings yet
- Mit8 323 s23 Pset 01solDocument9 pagesMit8 323 s23 Pset 01solAyham ziadNo ratings yet
- AERO 4630: Structural Dynamics Homework 5: 1 Problem 1: Viscously Damped PendulumDocument5 pagesAERO 4630: Structural Dynamics Homework 5: 1 Problem 1: Viscously Damped PendulumMD GOLAM SARWARNo ratings yet
- 5ESB0 2018 05 Midterm AnswersDocument5 pages5ESB0 2018 05 Midterm Answerscancer 123No ratings yet
- 4 5920319840068834062Document23 pages4 5920319840068834062samslimane6No ratings yet
- Introduction To Communication Systems 1st Edition Madhow Solutions ManualDocument18 pagesIntroduction To Communication Systems 1st Edition Madhow Solutions Manualbradleygillespieditcebswrf100% (15)
- Transient Conduction: The Lumped Capacitance MethodDocument29 pagesTransient Conduction: The Lumped Capacitance MethodJoão CelestinoNo ratings yet
- Dang 2020Document21 pagesDang 2020rahayu rizki putriNo ratings yet
- 9781107059320-SOLUTIONS Essential Digital SolutionsDocument47 pages9781107059320-SOLUTIONS Essential Digital SolutionsGulrez MNo ratings yet
- A Short Report On MATLAB Implementation of The Girsanov Transformation For Reliability AnalysisDocument8 pagesA Short Report On MATLAB Implementation of The Girsanov Transformation For Reliability AnalysisYoung EngineerNo ratings yet
- ME311 Autumn2021 T5Document5 pagesME311 Autumn2021 T5Varun PathakNo ratings yet
- Lagrangian InterpolationDocument10 pagesLagrangian InterpolationEnos Lolang100% (2)
- HW 1Document3 pagesHW 1Zero ChengNo ratings yet
- Homework Solution 4Document12 pagesHomework Solution 4Diego OrtizNo ratings yet
- Numerical Methods For Differential Equations: Euler MethodDocument27 pagesNumerical Methods For Differential Equations: Euler Methodmicky kololuNo ratings yet
- Report 6Document4 pagesReport 6Đình TrườngNo ratings yet
- Chapter 2. Representation of Signals and SystemsDocument44 pagesChapter 2. Representation of Signals and SystemsSuad KasapovićNo ratings yet
- Student Lecture 35 Inverse Laplace Transforms and U (T-A)Document7 pagesStudent Lecture 35 Inverse Laplace Transforms and U (T-A)uploadingpersonNo ratings yet
- r2 GoeDocument4 pagesr2 GoeBitan kumar DeNo ratings yet
- r2 PoissonDocument4 pagesr2 PoissonBitan kumar DeNo ratings yet
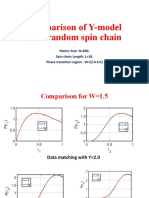
- Matching Gap Ratio With XXZ Spin ChainDocument25 pagesMatching Gap Ratio With XXZ Spin ChainBitan kumar DeNo ratings yet
- r4 GoeDocument4 pagesr4 GoeBitan kumar DeNo ratings yet
- r4 PoissonDocument4 pagesr4 PoissonBitan kumar DeNo ratings yet
- Miniproject-Advanced ArmDocument1 pageMiniproject-Advanced ArmBitan kumar DeNo ratings yet
- Izrailev 1990Document94 pagesIzrailev 1990Bitan kumar DeNo ratings yet
- r0 PoissonDocument4 pagesr0 PoissonBitan kumar DeNo ratings yet
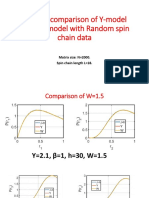
- Relative comparison of Y-model and β-h model with Random spin chain dataDocument19 pagesRelative comparison of Y-model and β-h model with Random spin chain dataBitan kumar DeNo ratings yet
- Quantum Transport in Gapped Graphene - 2019Document160 pagesQuantum Transport in Gapped Graphene - 2019Bitan kumar DeNo ratings yet
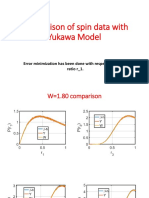
- Matching Spin-Data With Yukawa ModelDocument19 pagesMatching Spin-Data With Yukawa ModelBitan kumar DeNo ratings yet
- Hamiltonians of Quantum Dot-Based Systems: Single Quantum-Dot Coupled To Metallic Reser-Voirs and Phonon BathDocument2 pagesHamiltonians of Quantum Dot-Based Systems: Single Quantum-Dot Coupled To Metallic Reser-Voirs and Phonon BathBitan kumar DeNo ratings yet
- Comparison of Y-Model With Random Spin ChainDocument21 pagesComparison of Y-Model With Random Spin ChainBitan kumar DeNo ratings yet
- Matching Spin-Data With Yukawa ModelDocument19 pagesMatching Spin-Data With Yukawa ModelBitan kumar DeNo ratings yet
- Comparison of Spin Data With Yukawa Model: Error Minimization Has Been Done With Respect To The Gap Ratio R - 1Document13 pagesComparison of Spin Data With Yukawa Model: Error Minimization Has Been Done With Respect To The Gap Ratio R - 1Bitan kumar DeNo ratings yet
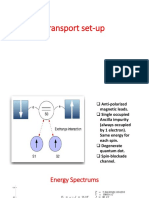
- Transport Set UpDocument5 pagesTransport Set UpBitan kumar DeNo ratings yet
- Comparison of Y-Model With Random Spin ChainDocument21 pagesComparison of Y-Model With Random Spin ChainBitan kumar DeNo ratings yet
- Theory of MagnetismDocument94 pagesTheory of MagnetismBitan kumar DeNo ratings yet
- Comparison of Y-Model With Random Spin ChainDocument21 pagesComparison of Y-Model With Random Spin ChainBitan kumar DeNo ratings yet
- Transport Set UpDocument5 pagesTransport Set UpBitan kumar DeNo ratings yet
- Nonequilibrium Green's Functions: Freely Taken Mainly From Rammer and Smith (1986) and Haug and Jauho (1998)Document35 pagesNonequilibrium Green's Functions: Freely Taken Mainly From Rammer and Smith (1986) and Haug and Jauho (1998)Bitan kumar DeNo ratings yet
- Quantum II Solved Question-2Document45 pagesQuantum II Solved Question-2Allizwell OSNo ratings yet
- Lecture 15Document5 pagesLecture 15JOSE CARLOS LEON GONZALEZNo ratings yet
- Exam VT2014Document4 pagesExam VT2014lolNo ratings yet
- The Electric Properties of Molecules: ExercisesDocument16 pagesThe Electric Properties of Molecules: ExercisesSergio Magalhaes FerreiraNo ratings yet
- Corrected Homework 5Document8 pagesCorrected Homework 5bruhbruhbruhNo ratings yet
- BSC SyllabusDocument42 pagesBSC SyllabussanjeevchsNo ratings yet
- Geophys. J. Int. 1989 Randall 469 81Document13 pagesGeophys. J. Int. 1989 Randall 469 81Mariano ArnaizNo ratings yet
- Non Equilibrium Greens Functions For Dummies Intr PDFDocument10 pagesNon Equilibrium Greens Functions For Dummies Intr PDFBhaskar KNo ratings yet
- Advanved Quantum Mechanics Exam Freie Universitat BerlinDocument3 pagesAdvanved Quantum Mechanics Exam Freie Universitat BerlinjuanNo ratings yet
- Models: 1, Condensations in Expanding CosmologicDocument3 pagesModels: 1, Condensations in Expanding CosmologicsayanNo ratings yet
- S. Zdravkovic, J.A. Tuszynski and M.V. Sataric: Peyrard-Bishop-Dauxois Model of DNA Dynamics and Impact of ViscosityDocument9 pagesS. Zdravkovic, J.A. Tuszynski and M.V. Sataric: Peyrard-Bishop-Dauxois Model of DNA Dynamics and Impact of ViscosityUloffNo ratings yet
- Foundations of Quantum MechanicsDocument35 pagesFoundations of Quantum MechanicsYi ChaiNo ratings yet
- Lecture 3Document18 pagesLecture 3بوبي بابيNo ratings yet
- M.SC Syllabus Astrophysics IVDocument31 pagesM.SC Syllabus Astrophysics IVPiyush BoseNo ratings yet
- Accuracy of Stochastic Perturbation Methods: The Case of Asset Pricing ModelsDocument21 pagesAccuracy of Stochastic Perturbation Methods: The Case of Asset Pricing ModelsJeremey Julian PonrajahNo ratings yet
- Introduction To Singular Perturbation Theory: Erika MayDocument24 pagesIntroduction To Singular Perturbation Theory: Erika MaybappaNo ratings yet
- The exact: C-function in integrable λ-deformed theoriesDocument16 pagesThe exact: C-function in integrable λ-deformed theoriesJack Ignacio NahmíasNo ratings yet
- Exercise 3Document2 pagesExercise 3Max JenkinsonNo ratings yet
- MSCPH 02Document7 pagesMSCPH 02Mahavir MoondNo ratings yet
- Phase PortraitsDocument39 pagesPhase PortraitsShivamNo ratings yet
- Homework 2: #'S 5.1, 5.3, 5.4, 5.7, 5.8, 5.9, 5.11: Michael Good Feb 3, 2005Document15 pagesHomework 2: #'S 5.1, 5.3, 5.4, 5.7, 5.8, 5.9, 5.11: Michael Good Feb 3, 2005Ioana Cristina GhineaNo ratings yet
- Solved Problems in Quantum MechanicsDocument3 pagesSolved Problems in Quantum MechanicsMalvado Aun Mas MalvadoNo ratings yet
- Anharmonic Oscillator in Quantum MechanicsDocument7 pagesAnharmonic Oscillator in Quantum MechanicsRobert RingstadNo ratings yet
- Zubov1997 PDFDocument12 pagesZubov1997 PDFwintersoulsNo ratings yet
- QFT Notes: Sarthak DuaryDocument7 pagesQFT Notes: Sarthak DuarySarthakNo ratings yet
- Principles of Physical ChemistryDocument23 pagesPrinciples of Physical ChemistryShibu Kumard0% (1)
- Physics SyllabusDocument11 pagesPhysics SyllabusJustin BrockNo ratings yet
- Applied Mathematics and Computation: Solomon Manukure, Wen-Xiu MaDocument9 pagesApplied Mathematics and Computation: Solomon Manukure, Wen-Xiu MaAnonymous BFlqdMFKLNo ratings yet