You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5819)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (845)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (348)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Molecular Dynamics: Printed BookDocument1 pageMolecular Dynamics: Printed BookJamal JacksonNo ratings yet
- Introduction To Data Communication and Networking: Digital TransmissionDocument32 pagesIntroduction To Data Communication and Networking: Digital TransmissionJeon JungraeNo ratings yet
- Data Representation WorkbookDocument8 pagesData Representation WorkbookMonkeyDluffyNo ratings yet
- Introduction To The Theory of Computation-L1Document5 pagesIntroduction To The Theory of Computation-L1Mukesh Kumar SinghNo ratings yet
- Speed Control of DC Motor PDFDocument78 pagesSpeed Control of DC Motor PDFLuis Eduardo RibeiroNo ratings yet
- Secured Cloud Data Migration Technique by Competent Probabilistic Public Key EncryptionDocument23 pagesSecured Cloud Data Migration Technique by Competent Probabilistic Public Key EncryptionAzmi AbdulbaqiNo ratings yet
- An Overview of Bayesian EconometricsDocument30 pagesAn Overview of Bayesian Econometrics6doitNo ratings yet
- PID Gain KPDocument4 pagesPID Gain KPmohanNo ratings yet
- (Applied Condition Monitoring 15) Alfonso Fernandez Del Rincon, Fernando Viadero - Part19Document10 pages(Applied Condition Monitoring 15) Alfonso Fernandez Del Rincon, Fernando Viadero - Part19Uma TamilNo ratings yet
- Document 770Document6 pagesDocument 770thamthoiNo ratings yet
- Marius Muja: Tabletop Object DetectionDocument17 pagesMarius Muja: Tabletop Object DetectionWillow GarageNo ratings yet
- AI Intern ResumeDocument1 pageAI Intern ResumeAmeer HamzaNo ratings yet
- Test Bank Questions Chapter 6Document3 pagesTest Bank Questions Chapter 6Anonymous 8ooQmMoNs1No ratings yet
- Journal of Natural Gas Science and Engineering: Chiranth Hegde, K.E. GrayDocument9 pagesJournal of Natural Gas Science and Engineering: Chiranth Hegde, K.E. GrayVimal RajNo ratings yet
- Iterative Figure-Ground DiscriminationDocument4 pagesIterative Figure-Ground Discriminationmehdi13622473No ratings yet
- Python Command Explanation: Ae2235-I Aerospace Systems and Control Theory Command SummaryDocument2 pagesPython Command Explanation: Ae2235-I Aerospace Systems and Control Theory Command SummaryPythonraptorNo ratings yet
- FEMII LAB Buckling Analysis v2 PDFDocument6 pagesFEMII LAB Buckling Analysis v2 PDFOlivier GouveiaNo ratings yet
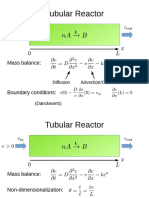
- Tubular Reactor: Mass BalanceDocument21 pagesTubular Reactor: Mass BalanceyohannesNo ratings yet
- Biostatistics MCQs 2024Document6 pagesBiostatistics MCQs 2024funnymemesworld2023No ratings yet
- MATLAB Based PCM Modeling and SimulationDocument7 pagesMATLAB Based PCM Modeling and SimulationPawan SinghNo ratings yet
- Spectral Analysis of SignalsDocument0 pagesSpectral Analysis of Signalssouvik5000No ratings yet
- Review - 3 - Load Forecasting PDFDocument25 pagesReview - 3 - Load Forecasting PDFhabte gebreial shrashrNo ratings yet
- Open SSLDocument31 pagesOpen SSLDibya SachiNo ratings yet
- Weather Trends Sales PredictorDocument6 pagesWeather Trends Sales PredictorWananda Muhammad RifkiNo ratings yet
- Experiment 5: Aim: To Implement Mc-Culloch and Pitt's Model For A Problem. TheoryDocument7 pagesExperiment 5: Aim: To Implement Mc-Culloch and Pitt's Model For A Problem. TheoryNidhi MhatreNo ratings yet
- Assignment 4Document4 pagesAssignment 4bot khanNo ratings yet
- BEE 4413 Exercise 1Document4 pagesBEE 4413 Exercise 1Hafzn HashimNo ratings yet
- Sem - III Business Mathematics Yearly PaperDocument12 pagesSem - III Business Mathematics Yearly Paperoc698067No ratings yet
- TOC 2076 - Ankit PangeniDocument21 pagesTOC 2076 - Ankit Pangenisheham ihjamNo ratings yet
- REGNO:311119106029 Melodina Carnelian D Loyola - Icam College of Engineering and Technology (Licet)Document6 pagesREGNO:311119106029 Melodina Carnelian D Loyola - Icam College of Engineering and Technology (Licet)MELODINA CARNELIAN 19EC061No ratings yet