You might also like
- Genetics Lab Exercise 10 - The Tools of DNA TechnologyDocument7 pagesGenetics Lab Exercise 10 - The Tools of DNA Technologybry uyNo ratings yet
- MIT Exam 2Document12 pagesMIT Exam 2Daniela Sanclemente100% (1)
- Critical Analysis For Psych 100 QueensDocument18 pagesCritical Analysis For Psych 100 QueensChloe AndrewsNo ratings yet
- Cho-K1 Ox2Document4 pagesCho-K1 Ox2valikshherbNo ratings yet
- Modern Data Mining Quiz 3Document5 pagesModern Data Mining Quiz 3NaynuhzNo ratings yet
- The Case of Thetainted" Taco ShellsDocument7 pagesThe Case of Thetainted" Taco ShellsDayakar PhotosNo ratings yet
- Lameness - Diagnostic TestsDocument12 pagesLameness - Diagnostic TestsS. Bala DahiyaNo ratings yet
- Basic Concepts of Data Mining, Clustering and Genetic AlgorithmsDocument26 pagesBasic Concepts of Data Mining, Clustering and Genetic AlgorithmsharisxyzNo ratings yet
- Luteinizing Hormone ReceptorDocument3 pagesLuteinizing Hormone ReceptorMuhammad Nurfajri JihaduddinNo ratings yet
- Section 4 Cladistics2Document5 pagesSection 4 Cladistics2Poohbearrr :No ratings yet
- Le Tuan Thanh-23BI14405Document10 pagesLe Tuan Thanh-23BI14405tl13032005No ratings yet
- Genetic AlgorithmDocument40 pagesGenetic AlgorithmGarima Singhal100% (1)
- Lovely Professional: Topic:Western BlottingDocument16 pagesLovely Professional: Topic:Western BlottingVikal RajputNo ratings yet
- Ensembl Exercises on Panda, Zebrafish, Mosquitoes, Bacteria, Human Genes & GenomeDocument6 pagesEnsembl Exercises on Panda, Zebrafish, Mosquitoes, Bacteria, Human Genes & Genome01 RerollNo ratings yet
- LAB 8 Chromosomal Abnormalities Gene Regulation 3Document4 pagesLAB 8 Chromosomal Abnormalities Gene Regulation 3Christina NgwakoNo ratings yet
- Mastering Biology Chapter 1Document22 pagesMastering Biology Chapter 1y95% (20)
- Lab 2 ds180 Genotyping LabDocument7 pagesLab 2 ds180 Genotyping Labapi-342081300No ratings yet
- AI-ML ProjectDocument17 pagesAI-ML ProjectJatin SehrawatNo ratings yet
- Hoechst 33342 HSC Staining and Stem Cell Purification ProtocolDocument7 pagesHoechst 33342 HSC Staining and Stem Cell Purification ProtocolbhawksNo ratings yet
- Science QuizDocument5 pagesScience QuizGaius Aurelius Valerius DiocletianusNo ratings yet
- Genome Browser ExerciseDocument5 pagesGenome Browser ExerciseyukiNo ratings yet
- Bioinformaticpdf 1Document21 pagesBioinformaticpdf 1melkamu genetNo ratings yet
- PhysioEx Exercise 3 Activity 1Document8 pagesPhysioEx Exercise 3 Activity 1tasyaauliatabina1122No ratings yet
- Tet-Off Advanced Inducible Gene Expression Systems User ManualDocument19 pagesTet-Off Advanced Inducible Gene Expression Systems User ManualJessica Asitimbay ZuritaNo ratings yet
- Sestrada Logistic Regression in R 02172023Document25 pagesSestrada Logistic Regression in R 02172023Mehdi Ben HammiNo ratings yet
- Squid Lab Packet v3Document8 pagesSquid Lab Packet v3Bradley Chung100% (1)
- PHD Thesis NeuroscienceDocument42 pagesPHD Thesis NeuroscienceAnil Bheemaiah100% (1)
- Western Blotting PDFDocument17 pagesWestern Blotting PDFAna Dominique EspiaNo ratings yet
- 6Document9 pages6Andreah BaylonNo ratings yet
- Genetic Algorithm BasicsDocument4 pagesGenetic Algorithm BasicskartikeyasarmaNo ratings yet
- Understanding the Action Potential ThresholdDocument5 pagesUnderstanding the Action Potential ThresholdPavel MilenkovskiNo ratings yet
- Group Assignment AIDocument7 pagesGroup Assignment AIKirthi VasanNo ratings yet
- PhysioEx Lab Report on Resting Membrane PotentialDocument8 pagesPhysioEx Lab Report on Resting Membrane PotentialJenna Picard100% (1)
- Diferential Expression Analysis PDFDocument72 pagesDiferential Expression Analysis PDFDavid LinhNo ratings yet
- Restriction Enzyme Digestion AnalysisDocument6 pagesRestriction Enzyme Digestion AnalysisLloaana 12No ratings yet
- Current Generated Flows Through Clips Placed On The Earlobes Output Current Adjustable From 80 To 600 Microamperes Circuit DiagramDocument6 pagesCurrent Generated Flows Through Clips Placed On The Earlobes Output Current Adjustable From 80 To 600 Microamperes Circuit DiagramemiroNo ratings yet
- BI Prac1Document4 pagesBI Prac1Maha SabirNo ratings yet
- Supplementary Materials for "ME-Class: A DNA Methylation-Based Machine Learning Approach for Cell Type ClassificationDocument14 pagesSupplementary Materials for "ME-Class: A DNA Methylation-Based Machine Learning Approach for Cell Type ClassificationMattNo ratings yet
- Clinical OncologyDocument13 pagesClinical Oncologyapi-585462575No ratings yet
- CS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesDocument2 pagesCS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesSUNDAS FATIMANo ratings yet
- CS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesDocument2 pagesCS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesSUNDAS FATIMANo ratings yet
- CS444: BIO INFORMATICS BLASTDocument2 pagesCS444: BIO INFORMATICS BLASTSUNDAS FATIMANo ratings yet
- CS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesDocument2 pagesCS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesSUNDAS FATIMANo ratings yet
- CS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesDocument2 pagesCS444: BIO INFORMATICS (Lab 1 - Manual) Bioinformatics Databases and Key Online ResourcesSUNDAS FATIMANo ratings yet
- Genetic Algorithms: Optimization TechniquesDocument36 pagesGenetic Algorithms: Optimization TechniquesPoonam Dhankar KunduNo ratings yet
- Physioex Lab Report: Pre-Lab Quiz ResultsDocument5 pagesPhysioex Lab Report: Pre-Lab Quiz ResultsWilhelm Sánchez100% (1)
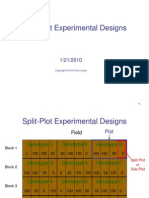
- Chapter9b. Split Plot Experiment 10april2011Document51 pagesChapter9b. Split Plot Experiment 10april2011Nor FaizahNo ratings yet
- Genetic AlgorithmDocument6 pagesGenetic AlgorithmShalinee Singh RathoreNo ratings yet
- Frog Muscle ProtocolDocument13 pagesFrog Muscle Protocolrjbagh08No ratings yet
- HBS 131 ResponseSheetDocument3 pagesHBS 131 ResponseSheetDelilah YacoubNo ratings yet
- Molecular Biology & Genetics: Essential Biology Self-Teaching GuideFrom EverandMolecular Biology & Genetics: Essential Biology Self-Teaching GuideNo ratings yet
- Nettle Extractions and BioassayDocument15 pagesNettle Extractions and BioassayAssignmentLab.comNo ratings yet
- Easy Differential Expression: F. Hahne and W. HuberDocument6 pagesEasy Differential Expression: F. Hahne and W. HuberulirschjNo ratings yet
- Winter Break Biotech Assignment 2012-13Document2 pagesWinter Break Biotech Assignment 2012-13Steven TruongNo ratings yet
- PhysioEx Lab Report on Resting Membrane PotentialDocument8 pagesPhysioEx Lab Report on Resting Membrane PotentialJoashan VacNo ratings yet
- Universal GenomeWalker 2.0 User Manual - 040314Document24 pagesUniversal GenomeWalker 2.0 User Manual - 040314xprakashNo ratings yet
- Logistic+Regression+Practice+Exercise+ +solutions - Ipynb ColaboratoryDocument5 pagesLogistic+Regression+Practice+Exercise+ +solutions - Ipynb ColaboratorySHEKHAR SWAMINo ratings yet
- Figure Above: Biological Inspiration of Genetic AlgorithmDocument6 pagesFigure Above: Biological Inspiration of Genetic AlgorithmPartha Pratim MazumderNo ratings yet
- Chapter 5: Modern Sequencing: - Sequencing by Mass SpectrometryDocument28 pagesChapter 5: Modern Sequencing: - Sequencing by Mass SpectrometryKyle BroflovskiNo ratings yet
- Ga02 Genetic Algorithms DemystifiedDocument8 pagesGa02 Genetic Algorithms DemystifiedIoachim DipseNo ratings yet
- Listening Sample Task - Matching Example 1Document4 pagesListening Sample Task - Matching Example 1Cristiane SerbanNo ratings yet
- QuestionDocument3 pagesQuestionFaisal ShahzadNo ratings yet
- Listening Sample Task - Plan Map Diagram LabellingDocument3 pagesListening Sample Task - Plan Map Diagram LabellingAdhi ThyanNo ratings yet
- Listening Sample Task - Form CompletionDocument4 pagesListening Sample Task - Form CompletionTrần Đăng Khoa100% (1)
- Listening - Sample - Task - Matching - Example - 2 - PDFDocument3 pagesListening - Sample - Task - Matching - Example - 2 - PDFSVS SudheerNo ratings yet
- Sequence Alignment/Map Format SpecificationDocument23 pagesSequence Alignment/Map Format SpecificationBinh VoNo ratings yet
- Awk Programming: 1. General 2. Awk With ConditionsDocument14 pagesAwk Programming: 1. General 2. Awk With ConditionsBinh VoNo ratings yet
- Unix Text ProcessingDocument680 pagesUnix Text Processingapi-3701136No ratings yet
- Nih Public Access: Bedtools: The Swiss-Army Tool For Genome Feature AnalysisDocument45 pagesNih Public Access: Bedtools: The Swiss-Army Tool For Genome Feature AnalysisBinh VoNo ratings yet
- PGD CNAD 607 Meal Management and Planning Disease SheetDocument14 pagesPGD CNAD 607 Meal Management and Planning Disease SheetHasaan QaziNo ratings yet
- Canadian Coding - ColonosDocument4 pagesCanadian Coding - ColonosFedrick BuenaventuraNo ratings yet
- Animal Models For Microbiome Research Advancing Basic andDocument115 pagesAnimal Models For Microbiome Research Advancing Basic andBioterio Funed100% (1)
- Soal PDFDocument113 pagesSoal PDFlimeddy100% (2)
- Studies On Design, Development and Characterization of Colon Targeted Drug Delivery of Mesalamine Using Coexcipient PolymerDocument7 pagesStudies On Design, Development and Characterization of Colon Targeted Drug Delivery of Mesalamine Using Coexcipient PolymerInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Gastroenterology and HepatologyDocument42 pagesGastroenterology and HepatologyCarlos HernándezNo ratings yet
- Breaking Down The Barriers: The Gut Microbiome, Intestinal Permeability and Stress-Related Psychiatric DisordersDocument20 pagesBreaking Down The Barriers: The Gut Microbiome, Intestinal Permeability and Stress-Related Psychiatric DisordersCSilva16No ratings yet
- Faecal Calprotectin: Review ArticleDocument14 pagesFaecal Calprotectin: Review Articletrisna amijayaNo ratings yet
- Colitis PDFDocument11 pagesColitis PDFicoanamareNo ratings yet
- Treatment of Ankylosing SpondylitisDocument16 pagesTreatment of Ankylosing SpondylitisrazvanbaranNo ratings yet
- Guidelines For Video Capsule Endoscopy: Emphasis On Crohn's DiseaseDocument8 pagesGuidelines For Video Capsule Endoscopy: Emphasis On Crohn's DiseaseNataliaMaedyNo ratings yet
- Water Kefir Handbook - Water Kefir Recipes, Step-By-Step Instructions, Health Benefits and More (Water Kefir Recipes, Water Kefir For Beginners, Fermented Drinks, Fermented Foods Book 1) (PDFDrive)Document72 pagesWater Kefir Handbook - Water Kefir Recipes, Step-By-Step Instructions, Health Benefits and More (Water Kefir Recipes, Water Kefir For Beginners, Fermented Drinks, Fermented Foods Book 1) (PDFDrive)angelobuffaloNo ratings yet
- Pathology Lab.: Gastrointestinal Tract MD IvDocument27 pagesPathology Lab.: Gastrointestinal Tract MD IvPablo SisirucaNo ratings yet
- Changes in Bowel HabitsDocument20 pagesChanges in Bowel HabitsSadaf SheikhNo ratings yet
- Lower Gastrointestinal BleedingDocument18 pagesLower Gastrointestinal Bleedingosama nawaysehNo ratings yet
- Choosing The Most Appropriate Biologic Therapy For Crohn's Disease According To Concomitant Extra-Intestinal Manifestations, Comorbidities, or Physiologic ConditionsDocument44 pagesChoosing The Most Appropriate Biologic Therapy For Crohn's Disease According To Concomitant Extra-Intestinal Manifestations, Comorbidities, or Physiologic ConditionsBernadett FarkasNo ratings yet
- Health and Wellness: Resource GuideDocument200 pagesHealth and Wellness: Resource GuideHaaroonNo ratings yet
- Nlex 1Document4 pagesNlex 1Aileen AlphaNo ratings yet
- DIARRHEADocument39 pagesDIARRHEAJessa RedondoNo ratings yet
- Engineering Probiotics For Therapeutic ApplicationsDocument9 pagesEngineering Probiotics For Therapeutic ApplicationsElinaNo ratings yet
- Diet in Benign Colonic Disorders: A Narrative ReviewDocument14 pagesDiet in Benign Colonic Disorders: A Narrative ReviewHouda LaatabiNo ratings yet
- Wedgewood Partners Second Quarter 2017 Client Letter ReviewDocument25 pagesWedgewood Partners Second Quarter 2017 Client Letter Reviewcalculatedrisk1No ratings yet
- AUF SOM - Dse of Small Intestine PDFDocument166 pagesAUF SOM - Dse of Small Intestine PDFYestin Reece Corpus ArcegaNo ratings yet
- Colorektal CancerDocument24 pagesColorektal CancerAgus PraktyasaNo ratings yet
- Nus Winning Markteing Pitch 2Document16 pagesNus Winning Markteing Pitch 2Vincent KohNo ratings yet
- Criterios de Elegibilidad de ACODocument88 pagesCriterios de Elegibilidad de ACOquimegiNo ratings yet
- BUHLMANN Diagnostics Corp. Announces New Distribution Agreement With Meridian Biosciences, Inc.Document3 pagesBUHLMANN Diagnostics Corp. Announces New Distribution Agreement With Meridian Biosciences, Inc.PR.comNo ratings yet
- Lower GI BleedingDocument40 pagesLower GI BleedingMohammad Firdaus100% (2)
- BR J Dermatol - 2022 - Dissemond - Inflammatory Skin Diseases and WoundsDocument11 pagesBR J Dermatol - 2022 - Dissemond - Inflammatory Skin Diseases and Woundsวสุชล ชัยชาญNo ratings yet
- Gastrointestinal Health Problems Test DrillDocument9 pagesGastrointestinal Health Problems Test DrillChristian Angel PoncianoNo ratings yet