You might also like
- Scientific Proof of ReincarnationDocument19 pagesScientific Proof of ReincarnationAsheesh MishraNo ratings yet
- Biotechnology and Genetic Engineering Summary PPT Igcse CaieDocument16 pagesBiotechnology and Genetic Engineering Summary PPT Igcse Caiearyan patelNo ratings yet
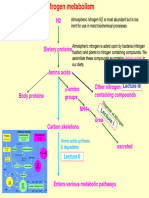
- Protein MetabolismDocument15 pagesProtein MetabolismAfshan Akhtar100% (1)
- Biochemistry Chapter 3Document5 pagesBiochemistry Chapter 3brownhazelNo ratings yet
- Key Topics: To Know: To Generate Energy by Acetyl Coa OxidationDocument23 pagesKey Topics: To Know: To Generate Energy by Acetyl Coa OxidationIsaiah Emmanuel SuguitanNo ratings yet
- Leukemia, Lymphoma Children PDFDocument31 pagesLeukemia, Lymphoma Children PDFPartha GanesanNo ratings yet
- Mesothelioma: A Clinical ReviewDocument43 pagesMesothelioma: A Clinical Reviewmynoidanh19No ratings yet
- Unit 9 Lipid MetabolismDocument28 pagesUnit 9 Lipid MetabolismAthina100% (1)
- Secondary Metabolism NotesDocument11 pagesSecondary Metabolism Notesleanne_tan_4No ratings yet
- 8 Abnormal BehaviorDocument34 pages8 Abnormal BehaviorRyanito MarquezNo ratings yet
- ch15 Organic ChemistryDocument117 pagesch15 Organic Chemistrybrownhazel50% (4)
- A2 Biology Edexcel Textbook Answers To QuestionsDocument56 pagesA2 Biology Edexcel Textbook Answers To Questionsbulanula63% (19)
- ch20 Organic Chemistry SolomonsDocument103 pagesch20 Organic Chemistry Solomonsbrownhazel78% (9)
- PROTEIN METABOLISM Dea Farha Fira Darson FineDocument44 pagesPROTEIN METABOLISM Dea Farha Fira Darson FineFarhati MardhiyahNo ratings yet
- ch13 ProblemsDocument84 pagesch13 Problemsbrownhazel67% (6)
- Metabolism of Lipids (2) NewDocument64 pagesMetabolism of Lipids (2) NewLyra Get100% (1)
- Herbs For PancreasDocument3 pagesHerbs For PancreasStefanie LevinaWolfNo ratings yet
- METABOLISME ASAM AMINO Protein BiologiDocument31 pagesMETABOLISME ASAM AMINO Protein BiologiAxzchiuu :vNo ratings yet
- Amino Acid MetabolismDocument15 pagesAmino Acid Metabolismshanto.tn98No ratings yet
- Amino Acid Metabolism All LecturesDocument18 pagesAmino Acid Metabolism All Lecturesmizare29gNo ratings yet
- Amino Acid MetabolismDocument10 pagesAmino Acid MetabolismatifzeaNo ratings yet
- BIOSINTESIS PROTlanjutanDocument31 pagesBIOSINTESIS PROTlanjutanAprilikkaearlyNo ratings yet
- C10 Protein and Amino Acid MetabolismDocument8 pagesC10 Protein and Amino Acid MetabolismSoraya D. Al-ObinayNo ratings yet
- Amino Acid Metabolism 2Document29 pagesAmino Acid Metabolism 2Francis GacheruNo ratings yet
- Chemistrry AllDocument7 pagesChemistrry AllTHARSHANA JERUSALEMNo ratings yet
- Protein, Nitrogen Katabolisme Dan Siklus UreaDocument35 pagesProtein, Nitrogen Katabolisme Dan Siklus UreaAnonymous QCMhA4wNgBNo ratings yet
- Amino Acids BiosynthesisDocument56 pagesAmino Acids BiosynthesisDeea LobonțiuNo ratings yet
- Protein MetabolismDocument6 pagesProtein MetabolismGandesa LangNo ratings yet
- Answers of Exam Ques.Document3 pagesAnswers of Exam Ques.Sonali SehrawatNo ratings yet
- Protein MetabolismDocument19 pagesProtein MetabolismAnn Michelle Tarrobago100% (1)
- Amino Acid Biosynthesis and DegradationDocument11 pagesAmino Acid Biosynthesis and DegradationFarhana AktharNo ratings yet
- 1.macronutrients Metabolism, interrelationship-HM 2019Document75 pages1.macronutrients Metabolism, interrelationship-HM 2019Denebo ErsuloNo ratings yet
- Protein Turnover and Amino Acid Catabolism: Chem 454: Biochemistry II University of Wisconsin-Eau ClaireDocument54 pagesProtein Turnover and Amino Acid Catabolism: Chem 454: Biochemistry II University of Wisconsin-Eau ClaireArif Mc TomsNo ratings yet
- Metabolism of Fatty AcidDocument40 pagesMetabolism of Fatty AcidjessicatieuuNo ratings yet
- Lecture 05. Lipid Metabolism-2. Lipogenesis. Metabolism of Cholesterol and Ketone Bodies.Document33 pagesLecture 05. Lipid Metabolism-2. Lipogenesis. Metabolism of Cholesterol and Ketone Bodies.Віталій Михайлович НечипорукNo ratings yet
- Protein and Amino Acid MetabolismDocument32 pagesProtein and Amino Acid MetabolismVirag0% (1)
- Metabolism of Essential AADocument7 pagesMetabolism of Essential AA48 Syeda KainatNo ratings yet
- Functions of GlycogenDocument11 pagesFunctions of GlycogenBea SamonteNo ratings yet
- Presented by "Prof. Dr. Masoom Khan" Lecture Notes. No. 678Document41 pagesPresented by "Prof. Dr. Masoom Khan" Lecture Notes. No. 678Qamar QamarNo ratings yet
- Amino Acid MetabolismDocument23 pagesAmino Acid MetabolismPRATEEK KUMARNo ratings yet
- كيمياء حيوية-1Document9 pagesكيمياء حيوية-1عائش العموديNo ratings yet
- Amino Acid Metabolism: The Nitrogen Cycle and Nitrogen FixationDocument54 pagesAmino Acid Metabolism: The Nitrogen Cycle and Nitrogen FixationAlbert TandyNo ratings yet
- CHAPTER 24 Lipid BiosynthesisDocument16 pagesCHAPTER 24 Lipid Biosynthesis楊畯凱No ratings yet
- Fatty Acids SynthesisDocument30 pagesFatty Acids SynthesisGhaidaa SadeqNo ratings yet
- Amino Acid Catabolism3Document2 pagesAmino Acid Catabolism3rolwinNo ratings yet
- Amino Acid Catabolism1Document5 pagesAmino Acid Catabolism1rolwinNo ratings yet
- Amino Acid CatabolismDocument57 pagesAmino Acid CatabolismMega Nur Hesti OktaviaNo ratings yet
- Amino Acid Metabolism 2Document97 pagesAmino Acid Metabolism 2EmmaNo ratings yet
- Amino Acid MetabolismDocument97 pagesAmino Acid MetabolismEmmaNo ratings yet
- 15BT103 Biochem-UNIT 3Document53 pages15BT103 Biochem-UNIT 3Adityanair RA1711009010128No ratings yet
- Microbial Physiology. Microbial Metabolism. Enzymes. Nutrition. Bioenergetics. Bacterial Growth and MultiplicationDocument102 pagesMicrobial Physiology. Microbial Metabolism. Enzymes. Nutrition. Bioenergetics. Bacterial Growth and MultiplicationamitNo ratings yet
- 9a. Metabolisme Asam AminoDocument62 pages9a. Metabolisme Asam AminohimawarumNo ratings yet
- Amino Acid DegradationDocument44 pagesAmino Acid DegradationTanvi ShindeNo ratings yet
- Pathways in Amino Acids and Protein Metabolism 2Document76 pagesPathways in Amino Acids and Protein Metabolism 2godfreybwerere13No ratings yet
- Protein MetaboilisamDocument18 pagesProtein MetaboilisamSumit PandyaNo ratings yet
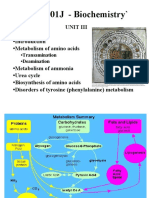
- Biochemistry Unit 03 PDFDocument66 pagesBiochemistry Unit 03 PDFDhanush kannanNo ratings yet
- Amino Acid Catabolism II: Dr. Mohammad AkramDocument19 pagesAmino Acid Catabolism II: Dr. Mohammad Akramapi-19824406No ratings yet
- Subjective QuestionsDocument5 pagesSubjective QuestionsAgustina MandasariNo ratings yet
- All 20 of The Amino Acids Present in Proteins Are Essential For HealthDocument9 pagesAll 20 of The Amino Acids Present in Proteins Are Essential For HealthDeepu ChaurasiyaNo ratings yet
- Biosynthetic Pathways - GPDocument46 pagesBiosynthetic Pathways - GPGhanshyam R ParmarNo ratings yet
- Medical Biochemistry (Week-15)Document5 pagesMedical Biochemistry (Week-15)wasimsafdarNo ratings yet
- Amino Acid DegredationDocument18 pagesAmino Acid DegredationMaxamed Faarax XaashiNo ratings yet
- Biosintesis Asam Lemak 1Document32 pagesBiosintesis Asam Lemak 1YainPanggaloNo ratings yet
- Biochemistry Homework HelpDocument10 pagesBiochemistry Homework HelpassignmentpediaNo ratings yet
- Fatty Acid Oxidation 312Document60 pagesFatty Acid Oxidation 312Ahmad LukmanNo ratings yet
- Introduction Amino Acid Matabolism and CatabolismDocument45 pagesIntroduction Amino Acid Matabolism and CatabolismAboubakar Moalim Mahad moh'd100% (1)
- Lect1 - 2017Document28 pagesLect1 - 2017George MakoriNo ratings yet
- Lipid MetabolismDocument94 pagesLipid MetabolismMiki MausNo ratings yet
- Biosynthesis of Fatty AcidsDocument3 pagesBiosynthesis of Fatty AcidsAhsan AliNo ratings yet
- Amino Acid Metabolism: Inborn ErrorsDocument18 pagesAmino Acid Metabolism: Inborn ErrorsAdedoyin BankoleNo ratings yet
- ch16 ProblemsDocument104 pagesch16 Problemsbrownhazel92% (12)
- Extractions 09 Acid BaseDocument9 pagesExtractions 09 Acid BasebrownhazelNo ratings yet
- Chapter 3 Water and Life: Campbell's Biology, 9e (Reece Et Al.)Document23 pagesChapter 3 Water and Life: Campbell's Biology, 9e (Reece Et Al.)brownhazelNo ratings yet
- Experiment #4: Acid/Base Extraction: ObjectivesDocument4 pagesExperiment #4: Acid/Base Extraction: ObjectivesbrownhazelNo ratings yet
- Atoms, Molecules, and Ions: - Atoms - Periodic Table - Chemical FormulaDocument44 pagesAtoms, Molecules, and Ions: - Atoms - Periodic Table - Chemical FormulabrownhazelNo ratings yet
- Urea CycleDocument22 pagesUrea CycleDawlat SalamaNo ratings yet
- TXTBK Yr1 1011Document3 pagesTXTBK Yr1 1011thuyduongenterNo ratings yet
- Special Articles Serum Albumin: Marcus Murray Oratz Sidney S. SchreiberDocument17 pagesSpecial Articles Serum Albumin: Marcus Murray Oratz Sidney S. SchreiberTuấn AnhNo ratings yet
- Circulatory System SEDocument4 pagesCirculatory System SEOMAR ID-AGRAM100% (1)
- Role of Mutation in Plant BreedingDocument9 pagesRole of Mutation in Plant BreedingBrijesh Kumar100% (1)
- MCQSDocument258 pagesMCQSmuhannedNo ratings yet
- Biology Zoology Botany MCQ PDFDocument27 pagesBiology Zoology Botany MCQ PDFAnonymous jxm0WNS7QaNo ratings yet
- 06 Notes Non-Mendelian Genetics Student-1Document20 pages06 Notes Non-Mendelian Genetics Student-1Ishaan SinghNo ratings yet
- Genetics Lessons 1 - 6Document20 pagesGenetics Lessons 1 - 6Suiee GenrieeNo ratings yet
- Kumpulan Soal Transfusi DarahDocument6 pagesKumpulan Soal Transfusi DarahdrnyolNo ratings yet
- EDUC 104 Report (Nik&Jeh)Document48 pagesEDUC 104 Report (Nik&Jeh)Nikki DanaNo ratings yet
- Exercises 5Document1 pageExercises 5Jonathhan RecaldeNo ratings yet
- Economic Importance of BacteriaDocument3 pagesEconomic Importance of Bacteriabiswajeet panda100% (1)
- S.y.bsc. Biotechnology (BT 211 & 212) Question BankDocument24 pagesS.y.bsc. Biotechnology (BT 211 & 212) Question BankShahab SaqibNo ratings yet
- 23650Document25 pages23650Ade FeriyatnaNo ratings yet
- Aleurone LayerDocument5 pagesAleurone LayerMian RashidNo ratings yet
- In Ammation and Tumor Progression: Signaling Pathways and Targeted InterventionDocument46 pagesIn Ammation and Tumor Progression: Signaling Pathways and Targeted Interventionenkiyna archNo ratings yet
- (Microbiology) Microbiological Food PoisoningDocument72 pages(Microbiology) Microbiological Food Poisoningamin100% (1)
- For and Against EssayDocument1 pageFor and Against EssayGame PlayerNo ratings yet
- Antigen Processing and Presentation PDFDocument36 pagesAntigen Processing and Presentation PDFMarinero CzarNo ratings yet
- Biology and Data Interpretation Techniques ConceptsDocument2 pagesBiology and Data Interpretation Techniques ConceptsVani DarjiNo ratings yet
- Case Study On GMOsDocument11 pagesCase Study On GMOsroselle azucena100% (1)
- Simultaneous Occurrence of Hypodontia and Microdontia in A Single CaseDocument3 pagesSimultaneous Occurrence of Hypodontia and Microdontia in A Single CaseNadeemNo ratings yet