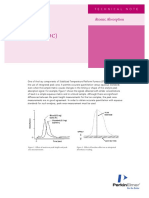
You might also like
- An Introduction To Inductively Coupled Plasma Mass Spectroscopy (ICP-MS) and Its ApplicationsDocument9 pagesAn Introduction To Inductively Coupled Plasma Mass Spectroscopy (ICP-MS) and Its ApplicationsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Rasayan Journal of ChemistryDocument7 pagesRasayan Journal of ChemistryAli KhumaeniNo ratings yet
- Comparación ICP-AAS VarianDocument7 pagesComparación ICP-AAS VarianVictoria OrtegaNo ratings yet
- ICP-OES ICP-MS and AAS Techniques ComparedDocument11 pagesICP-OES ICP-MS and AAS Techniques ComparedMuhammet Samet BülbülNo ratings yet
- ICP-MS - ICP-MS Instrumentation, ICP-MS Analysis, Strengths and LimitationDocument9 pagesICP-MS - ICP-MS Instrumentation, ICP-MS Analysis, Strengths and LimitationHd NsNo ratings yet
- SpectrometryDocument11 pagesSpectrometryJack StoneNo ratings yet
- Muhammad Ali Akbar - Resume ICP OES Dan ICP MSDocument5 pagesMuhammad Ali Akbar - Resume ICP OES Dan ICP MSrifqi bambangNo ratings yet
- ICP-MS Primer Encyc Anal Sci 2019Document9 pagesICP-MS Primer Encyc Anal Sci 2019Christopher GrahamNo ratings yet
- Minerals in WaterDocument7 pagesMinerals in WaterK JNo ratings yet
- Inductively Coupled PlasmaDocument8 pagesInductively Coupled Plasmagerarjui100% (1)
- Icp Ms ThesisDocument6 pagesIcp Ms Thesisdnrssvyt100% (2)
- Icp - OesDocument19 pagesIcp - OesLe Hong Tuan KietNo ratings yet
- Atomic Emission SpectrosDocument16 pagesAtomic Emission SpectrosHamza Ali MinhasNo ratings yet
- Unit 10 Atomic Emission SpectrometryDocument26 pagesUnit 10 Atomic Emission SpectrometryVelpuri Venkatappaiah67% (3)
- Icp AppendixDocument55 pagesIcp AppendixKarthikeyan JagannathanNo ratings yet
- Surface Plasmon Resonance Master ThesisDocument4 pagesSurface Plasmon Resonance Master Thesisbk34np00100% (2)
- 199 Manuscript - 329 1 10 20201016Document9 pages199 Manuscript - 329 1 10 20201016songyang hanNo ratings yet
- Icp MSDocument8 pagesIcp MSSa Nhsa100% (1)
- Highly Sensitive Immunoassay Based On Immunogold - Silver Amplification and Inductively Coupled Plasma Mass Spectrometric DetectionDocument7 pagesHighly Sensitive Immunoassay Based On Immunogold - Silver Amplification and Inductively Coupled Plasma Mass Spectrometric DetectionLylia MalakNo ratings yet
- Houk 1980Document7 pagesHouk 1980cjimeneztrianaNo ratings yet
- Going Bad: Icp-Oes Analysis of Metals in Apples: Kaitlyn MchughDocument13 pagesGoing Bad: Icp-Oes Analysis of Metals in Apples: Kaitlyn MchughKaitlyn McHughNo ratings yet
- Research Paper On Mass SpectrometryDocument10 pagesResearch Paper On Mass Spectrometryrpmvtcrif100% (1)
- Aes Lecture NoteDocument5 pagesAes Lecture NoteEmmanuella OffiongNo ratings yet
- Inductively Coupled Plasma Atomic Emission SpectrosDocument3 pagesInductively Coupled Plasma Atomic Emission SpectrosLihini NimsaraNo ratings yet
- U-Pb Zircon Dating by Laser ablation-MC-ICP-MS Using A New Multiple Ion Counting Faraday Collector ArrayDocument10 pagesU-Pb Zircon Dating by Laser ablation-MC-ICP-MS Using A New Multiple Ion Counting Faraday Collector ArrayjcpiedrahitavNo ratings yet
- Isotope-Ratio Mass SpectrometryDocument6 pagesIsotope-Ratio Mass SpectrometryEgie WijaksonoNo ratings yet
- Icp MSDocument68 pagesIcp MSluongthuong100% (1)
- CRPaptamer 1Document7 pagesCRPaptamer 1priyaNo ratings yet
- Instrumentation, Fundamentals, and Application of Laser Ablation-Inductively Coupled Plasma-Mass SpectrometryDocument19 pagesInstrumentation, Fundamentals, and Application of Laser Ablation-Inductively Coupled Plasma-Mass SpectrometryÁngelNo ratings yet
- Mass SpectrosDocument19 pagesMass SpectrosRadi RamansyahNo ratings yet
- LC Ms Training Protocol 2Document26 pagesLC Ms Training Protocol 2Hóa Học K64 Tài NăngNo ratings yet
- ICP-MS, or ICP-AES and AAS?-a ComparisonDocument7 pagesICP-MS, or ICP-AES and AAS?-a ComparisonNasReen Ibrahim Arif JamalNo ratings yet
- CHM026 - Exam 2 Part IDocument2 pagesCHM026 - Exam 2 Part Ijoseph cyron solidumNo ratings yet
- Localized Surface Plasmon Resonance ThesisDocument6 pagesLocalized Surface Plasmon Resonance ThesisCheryl Brown100% (2)
- Talanta 174 (2017) 652-659Document8 pagesTalanta 174 (2017) 652-659Luís CerdeiraNo ratings yet
- ICP Vs AASDocument6 pagesICP Vs AASĐầm Già Xì XìNo ratings yet
- Speciation of Six Arsenic Compounds Using Capillary Electrophoresis - Inductively Coupled Plasma Mass SpectrometryDocument6 pagesSpeciation of Six Arsenic Compounds Using Capillary Electrophoresis - Inductively Coupled Plasma Mass SpectrometryRoni Adi WijayaNo ratings yet
- Journal Pre-ProofsDocument37 pagesJournal Pre-ProofsÁLVARO CARLOS AGUADO MALLQUINo ratings yet
- Optical and Langmuir Probe Diagnostics IDocument6 pagesOptical and Langmuir Probe Diagnostics IJulia SyNo ratings yet
- Determination of A Brass Alloy Concentration Composition Using Calibration-Free Laser-Induced Breakdown SpectrosDocument12 pagesDetermination of A Brass Alloy Concentration Composition Using Calibration-Free Laser-Induced Breakdown SpectrosogonzalesrNo ratings yet
- First JurnalDocument5 pagesFirst JurnaljaneNo ratings yet
- Tatro M.E. - Optical Emission Inductively Coupled Plasma in Environmental AnalysisDocument7 pagesTatro M.E. - Optical Emission Inductively Coupled Plasma in Environmental AnalysisvladislavNo ratings yet
- Optical Diagnosis and Theoretical Simulation of Laser Induced Lead Plasma SpectrumDocument7 pagesOptical Diagnosis and Theoretical Simulation of Laser Induced Lead Plasma SpectrumHOD PhysicsNo ratings yet
- Neutron Activation Analysis (INAA and RNAA)Document4 pagesNeutron Activation Analysis (INAA and RNAA)Marco Antonio Enriquez QuirogaNo ratings yet
- AAS or ICP-OES-Are They Competing TechniquesDocument8 pagesAAS or ICP-OES-Are They Competing TechniquesKim HiềnNo ratings yet
- T. Lapainis Et Al - A Multichannel Native Fluorescence Detection System For Capillary Electrophoretic Analysis of Neurotransmitters in Single NeuronsDocument9 pagesT. Lapainis Et Al - A Multichannel Native Fluorescence Detection System For Capillary Electrophoretic Analysis of Neurotransmitters in Single NeuronsSour60No ratings yet
- Anion GapDocument13 pagesAnion GapLuis Gerardo Alcalá GonzálezNo ratings yet
- XX 44465 Icp Ms Elemental Guide Method Development Xx44465 enDocument42 pagesXX 44465 Icp Ms Elemental Guide Method Development Xx44465 enReem MohamedNo ratings yet
- A Selective Strategy For Determination of Ascorbic Acid Based On MolecularDocument6 pagesA Selective Strategy For Determination of Ascorbic Acid Based On MolecularSonyanurizkiNo ratings yet
- Langmuir Probe Diagnostics of Plasma in High CurrentDocument13 pagesLangmuir Probe Diagnostics of Plasma in High CurrentdeveloperstrustNo ratings yet
- Notes On LC-MSDocument6 pagesNotes On LC-MSAlabhya DasNo ratings yet
- A Comparison Between PIXE, ICP-AES and ICP-MSDocument6 pagesA Comparison Between PIXE, ICP-AES and ICP-MSLayth DrabeeNo ratings yet
- Unit 11 Applications of AAS and AESDocument22 pagesUnit 11 Applications of AAS and AESNathanian75% (4)
- Flame PhotoDocument5 pagesFlame PhotoAli Raza MalanaNo ratings yet
- Coronary Artery CTCalcium Score AsDocument12 pagesCoronary Artery CTCalcium Score AsJuan Lopez HernándezNo ratings yet
- Atomic SpectrosDocument28 pagesAtomic SpectrosJorge Rios RNo ratings yet
- Article ChemDocument16 pagesArticle ChemNyah SampangNo ratings yet
- Electrochemistry of Dihydroxybenzene Compounds: Carbon Based Electrodes and Their Uses in Synthesis and SensorsFrom EverandElectrochemistry of Dihydroxybenzene Compounds: Carbon Based Electrodes and Their Uses in Synthesis and SensorsNo ratings yet
- Quantitative Biological and Clinical Mass Spectrometry: An IntroductionFrom EverandQuantitative Biological and Clinical Mass Spectrometry: An IntroductionNo ratings yet
- Ion-Selective Electrode Reviews: Volume 6From EverandIon-Selective Electrode Reviews: Volume 6J. D. R. ThomasNo ratings yet
- AA Baseline Offset Correction Technical NoteDocument2 pagesAA Baseline Offset Correction Technical Noteونزار عبد القادرNo ratings yet
- AA Calibration Algorithms Technical NoteDocument2 pagesAA Calibration Algorithms Technical Noteونزار عبد القادرNo ratings yet
- 21 CFR Part 11 QuestionsDocument6 pages21 CFR Part 11 Questionsونزار عبد القادرNo ratings yet
- TP 01 Unit 03 Workbook Ak PDFDocument2 pagesTP 01 Unit 03 Workbook Ak PDFJimmy Castillo89% (9)
- Tabla para Espectroscopia DefinitivoDocument247 pagesTabla para Espectroscopia DefinitivoNicolas Rafael Meza SantiagoNo ratings yet
- 119 Phenolic Components by UV-Visible SpectraDocument3 pages119 Phenolic Components by UV-Visible Spectraونزار عبد القادرNo ratings yet
- Gravity Distribution Systems: A System Design and ConstructionDocument40 pagesGravity Distribution Systems: A System Design and ConstructionTooma DavidNo ratings yet
- INCOMPATIBILITIESDocument27 pagesINCOMPATIBILITIESArk Olfato Parojinog100% (1)
- English Solved SP3Document6 pagesEnglish Solved SP3Prem PatelNo ratings yet
- English 900 - 01Document147 pagesEnglish 900 - 01Hnin Hnin AungNo ratings yet
- Nonlinear Modeling of RC Structures Using Opensees: University of Naples Federico IiDocument66 pagesNonlinear Modeling of RC Structures Using Opensees: University of Naples Federico IiJorge Luis Garcia ZuñigaNo ratings yet
- Practice For Group Work Week 2 Case 2.1 - Assigning Plants To Products (Better Products Company)Document2 pagesPractice For Group Work Week 2 Case 2.1 - Assigning Plants To Products (Better Products Company)Thu TrangNo ratings yet
- ConferenceProceedings finalBalHNS2009Document332 pagesConferenceProceedings finalBalHNS2009kayron limaNo ratings yet
- Radiation Protection in Dental RadiologyDocument52 pagesRadiation Protection in Dental Radiologyivan dario ardila martinezNo ratings yet
- BUDGET OF WORK SY: 2021-2022: Subject: Tle 8 Grading Period: 3Document2 pagesBUDGET OF WORK SY: 2021-2022: Subject: Tle 8 Grading Period: 3michelle dayritNo ratings yet
- Sheree Hendersonresume 2Document3 pagesSheree Hendersonresume 2api-265774249No ratings yet
- VERITAS Cluster Server For UNIX, Fundamentals: Lesson 6 VCS Configuration MethodsDocument50 pagesVERITAS Cluster Server For UNIX, Fundamentals: Lesson 6 VCS Configuration MethodsNirmal AsaithambiNo ratings yet
- Natal Chart ReportDocument22 pagesNatal Chart ReportIngridNo ratings yet
- Project Work Class 11Document17 pagesProject Work Class 11DipeshNo ratings yet
- SM2 Polygon of ForcesDocument11 pagesSM2 Polygon of ForcesMel DNo ratings yet
- Arte PoveraDocument13 pagesArte PoveraSohini MaitiNo ratings yet
- Baylan: Water Meters With M-BusDocument2 pagesBaylan: Water Meters With M-Busamr ibrahimNo ratings yet
- AaaaaDocument3 pagesAaaaaAnonymous C3BD7OdNo ratings yet
- 003 Users Manuel of Safir 2016 - MechanicalDocument74 pages003 Users Manuel of Safir 2016 - Mechanicalenrico_britaiNo ratings yet
- NDX DolsonDocument67 pagesNDX DolsonMahanta BorahNo ratings yet
- ADM - DIASS 11 Q4 Weeks 1-8 - LatestDocument28 pagesADM - DIASS 11 Q4 Weeks 1-8 - LatestjoyceNo ratings yet
- Soal TPS Bahasa InggrisDocument3 pagesSoal TPS Bahasa InggrisMaya Putri EkasariNo ratings yet
- OLET1139 - Essay - July 21 - InstructionDocument3 pagesOLET1139 - Essay - July 21 - InstructionPriscaNo ratings yet
- We Exist As Molecular Structures.: Have You Found Your Path?Document9 pagesWe Exist As Molecular Structures.: Have You Found Your Path?Stephen KingNo ratings yet
- Computed Radiography 1Document38 pagesComputed Radiography 1Charisa Antonette HuelvaNo ratings yet
- Case StudyDocument11 pagesCase StudyCyril CauilanNo ratings yet
- Astm D 2979 PDFDocument3 pagesAstm D 2979 PDFMiftahulArifNo ratings yet
- Soil PHDocument19 pagesSoil PHElly Paul Andres TomasNo ratings yet
- StringDocument4 pagesStringAadyant BhadauriaNo ratings yet
- Ministry of Health and Family Welfare Pregnancy GuidelinesDocument173 pagesMinistry of Health and Family Welfare Pregnancy GuidelinesKhushi GuptaNo ratings yet
- 29 PerformanceAssessmentDocument22 pages29 PerformanceAssessmentDarmanNo ratings yet