You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (589)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5806)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Mescaline MakingDocument2 pagesMescaline MakingJohnyfrostNo ratings yet
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Organic Chemistry - Reactions and MechanismsDocument120 pagesOrganic Chemistry - Reactions and MechanismsLoveena Steadman100% (8)
- Hydrodesulfurization of Dibenzothiophene On A Como/Al O Catalyst: Reaction Network and KineticsDocument8 pagesHydrodesulfurization of Dibenzothiophene On A Como/Al O Catalyst: Reaction Network and KineticsGrupo de Investigaciones en CatalisisNo ratings yet
- 0273e Fatty Alcohol EsterDocument6 pages0273e Fatty Alcohol Esterhada33100% (1)
- Chemical Engineering October 2013Document76 pagesChemical Engineering October 2013Hani KirmaniNo ratings yet
- Applied Homogeneous Catalysis With Organo-Metallic Compounds - 2nd EditionDocument1,492 pagesApplied Homogeneous Catalysis With Organo-Metallic Compounds - 2nd Editionharrypoutreur100% (4)
- Ou 2015Document7 pagesOu 2015Luis M. MolinaNo ratings yet
- The Quantum Sutton-Chen Many-Body Potential For PRDocument31 pagesThe Quantum Sutton-Chen Many-Body Potential For PRLuis M. MolinaNo ratings yet
- ProgrammeDocument4 pagesProgrammeLuis M. MolinaNo ratings yet
- ChisteDocument2 pagesChisteLuis M. MolinaNo ratings yet
- WS 2012 SM1 Worksheet2Document11 pagesWS 2012 SM1 Worksheet2Luis M. MolinaNo ratings yet
- Sutton 1990Document9 pagesSutton 1990Luis M. MolinaNo ratings yet
- Aguado 2001Document9 pagesAguado 2001Luis M. MolinaNo ratings yet
- Nmat 2384Document4 pagesNmat 2384Luis M. MolinaNo ratings yet
- Aguado 1999Document11 pagesAguado 1999Luis M. MolinaNo ratings yet
- APS 2021 X10 Al2O3 CatalysisDocument15 pagesAPS 2021 X10 Al2O3 CatalysisLuis M. MolinaNo ratings yet
- CMS 2015 Pt3M CO OxidationDocument9 pagesCMS 2015 Pt3M CO OxidationLuis M. MolinaNo ratings yet
- Catalysis Ot ClustersDocument8 pagesCatalysis Ot ClustersLuis M. MolinaNo ratings yet
- Molecular Orbital of Chemisorbed Carbon Monoxide: GeohgeDocument6 pagesMolecular Orbital of Chemisorbed Carbon Monoxide: GeohgeLuis M. MolinaNo ratings yet
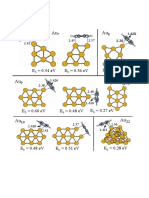
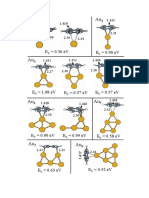
- Figure 2Document1 pageFigure 2Luis M. MolinaNo ratings yet
- Texlive en PDFDocument42 pagesTexlive en PDFLuis M. MolinaNo ratings yet
- AuN Plus Benz 1Document1 pageAuN Plus Benz 1Luis M. MolinaNo ratings yet
- Machine Learning For Interatomic Potential Models: Articles You May Be Interested inDocument14 pagesMachine Learning For Interatomic Potential Models: Articles You May Be Interested inLuis M. MolinaNo ratings yet
- Intercalation of Transition Metals Into Stacked Benzene Rings: A Model Study of The Intercalation of Transition Metals Into Bilayered GrapheneDocument7 pagesIntercalation of Transition Metals Into Stacked Benzene Rings: A Model Study of The Intercalation of Transition Metals Into Bilayered GrapheneLuis M. MolinaNo ratings yet
- Figure 1Document1 pageFigure 1Luis M. MolinaNo ratings yet
- DFT Study of Propene Epoxidation Mechanisms at Silver NanoparticlesDocument11 pagesDFT Study of Propene Epoxidation Mechanisms at Silver NanoparticlesLuis M. MolinaNo ratings yet
- Hydrogenation: Heterogeneous CatalysisDocument10 pagesHydrogenation: Heterogeneous CatalysisAnand SwarnkarNo ratings yet
- Reactive and Hybrid Separations: Incentives, Applications, BarriersDocument48 pagesReactive and Hybrid Separations: Incentives, Applications, BarriersKarime KantunNo ratings yet
- Alkyne ReactionsDocument3 pagesAlkyne ReactionsJudith de RoxasNo ratings yet
- Pembuatab KatalisDocument8 pagesPembuatab Katalisyusniya skNo ratings yet
- Principles of General Organic and Biological Chemistry 2Nd Edition Smith Test Bank Full Chapter PDFDocument46 pagesPrinciples of General Organic and Biological Chemistry 2Nd Edition Smith Test Bank Full Chapter PDFBrettClinewdjc100% (8)
- Hydrodesulfurization of Dibenzothiophene Over Ni-Mo-W Sulfide Catalysts Supported On Sol-Gel Al2O3-CeO2Document22 pagesHydrodesulfurization of Dibenzothiophene Over Ni-Mo-W Sulfide Catalysts Supported On Sol-Gel Al2O3-CeO2Hadis Shojaei0% (1)
- 54.1 Arenes Benzene Chemistry Ial Edexcel Chemistry QPDocument19 pages54.1 Arenes Benzene Chemistry Ial Edexcel Chemistry QPBooksNo ratings yet
- Surface Structure and Catalytic Activity of Sulfided Moo,/Al, O, Catalysts: Hydrodesulfurization and Hydrogenation ActivitiesDocument8 pagesSurface Structure and Catalytic Activity of Sulfided Moo,/Al, O, Catalysts: Hydrodesulfurization and Hydrogenation ActivitiesAhmad Edwin Al HasyirNo ratings yet
- Binepines, ChemSocRew2011 Gladiali ReviewDocument20 pagesBinepines, ChemSocRew2011 Gladiali ReviewszbaloghNo ratings yet
- ACS Appl. MAter. Interfaces, 2020, 12, 7285-7294Document10 pagesACS Appl. MAter. Interfaces, 2020, 12, 7285-7294Grad OanaNo ratings yet
- Hydrocarbons MarathonDocument353 pagesHydrocarbons MarathonGully GamingNo ratings yet
- Highly Selective Conversion of CO2 Into Ethanol On CuZnOAl2O3 Catalyst With The Assistance of PlasmaDocument6 pagesHighly Selective Conversion of CO2 Into Ethanol On CuZnOAl2O3 Catalyst With The Assistance of PlasmaNguyễn TuânNo ratings yet
- Processes 07 00136Document22 pagesProcesses 07 00136vanessa jimenezNo ratings yet
- Synthesis of P-Aminophenol by Catalytic Hydrogenation of P-NitrophenolDocument7 pagesSynthesis of P-Aminophenol by Catalytic Hydrogenation of P-NitrophenolFelipe TavaresNo ratings yet
- Elena Petricci and Maurizio Taddei - Microwave Assisted Reactions With Gas ReagentsDocument5 pagesElena Petricci and Maurizio Taddei - Microwave Assisted Reactions With Gas ReagentsnnnnjwNo ratings yet
- Chapter 16 Lecture NotesDocument15 pagesChapter 16 Lecture NotesJSGINo ratings yet
- Harshaw Catalysts: IftefïeiDocument1 pageHarshaw Catalysts: IftefïeiMustafa DemircioğluNo ratings yet
- ACCE 513 07 - Deactivation and Regenaration of CatalystsDocument11 pagesACCE 513 07 - Deactivation and Regenaration of CatalystsSufal BiswasNo ratings yet
- Amines BrochureDocument6 pagesAmines BrochureFRANCIS NDOURNo ratings yet
- Isooctane: Catalytic DistillationDocument12 pagesIsooctane: Catalytic DistillationAashish GauravNo ratings yet
- Unit-Ii: Sulfur and Natural ProductsDocument55 pagesUnit-Ii: Sulfur and Natural Productsomer faruqeNo ratings yet
- Lecture 11 Hydrogenation and Its ThermodynamicsDocument28 pagesLecture 11 Hydrogenation and Its ThermodynamicsHuraira AbidNo ratings yet
- Chemical Reactions of OilDocument51 pagesChemical Reactions of OilrosemagnaNo ratings yet
- Chapter 12 Chemical KineticsDocument90 pagesChapter 12 Chemical KineticsGurudutt SharmaNo ratings yet