You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (589)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5806)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- 2005 Heart+Disease+Diagnosis+and+TherapyDocument537 pages2005 Heart+Disease+Diagnosis+and+TherapyrominNo ratings yet
- Antenatal Info Booklet Intuitive BirthDocument32 pagesAntenatal Info Booklet Intuitive Birthapi-223713414No ratings yet
- PharmacotherapyDocument42 pagesPharmacotherapykhalid a.qazi100% (1)
- Atrial Septal Defect (ASD)Document35 pagesAtrial Septal Defect (ASD)Nur Arifah Astri100% (2)
- A Guide To ArthritisDocument12 pagesA Guide To ArthritisConsidraCareNo ratings yet
- Dornase Alfa in Cystic Fibrosis: Indications, Comparative Studies and Effects On Lung Clearance IndexDocument7 pagesDornase Alfa in Cystic Fibrosis: Indications, Comparative Studies and Effects On Lung Clearance IndexIván Gustavo Magaña CeballosNo ratings yet
- Pulmonary Hypertension in Bronchopulmonary DysplasiaDocument10 pagesPulmonary Hypertension in Bronchopulmonary DysplasiaIván Gustavo Magaña CeballosNo ratings yet
- Preventing Bronchopulmonary Dysplasia New Tools For An Old ChallengeDocument10 pagesPreventing Bronchopulmonary Dysplasia New Tools For An Old ChallengeIván Gustavo Magaña CeballosNo ratings yet
- Positive End-Expiratory Pressure For Preterm Infants Requiring Conventional Mechanical Ventilation For Respiratory Distress Syndrome or Bronchopulmonary DysplasiaDocument57 pagesPositive End-Expiratory Pressure For Preterm Infants Requiring Conventional Mechanical Ventilation For Respiratory Distress Syndrome or Bronchopulmonary DysplasiaIván Gustavo Magaña CeballosNo ratings yet
- Epidemiology and Risk Factors For Bronchopilmonary Dysplasia Less 32 WeeksDocument10 pagesEpidemiology and Risk Factors For Bronchopilmonary Dysplasia Less 32 WeeksIván Gustavo Magaña CeballosNo ratings yet
- Early 8 Days Systemic Postnatal Corticosteroids For Prevention of DBPDocument117 pagesEarly 8 Days Systemic Postnatal Corticosteroids For Prevention of DBPIván Gustavo Magaña CeballosNo ratings yet
- Broncoscopia 3Document1 pageBroncoscopia 3Iván Gustavo Magaña CeballosNo ratings yet
- GammaglobulinaDocument7 pagesGammaglobulinaIván Gustavo Magaña CeballosNo ratings yet
- Abraxane DetailDocument7 pagesAbraxane DetailGoel VaibhavNo ratings yet
- Chemical Burn 3Document4 pagesChemical Burn 3Yeni PuspitasariNo ratings yet
- Thyroid NodulesDocument7 pagesThyroid NodulesPravat SatpathyNo ratings yet
- אנדוDocument111 pagesאנדוLiav KfirNo ratings yet
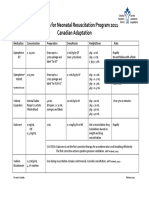
- Medications For Neonatal Resuscitation Program 2011 Canadian AdaptationDocument1 pageMedications For Neonatal Resuscitation Program 2011 Canadian AdaptationrubymayNo ratings yet
- PruBSN HEP+ BrochureDocument18 pagesPruBSN HEP+ BrochureMalik TaufiqNo ratings yet
- Dietary Supplements in Dermatology: A Review of The Evidence For Zinc, Biotin, Vitamin D, Nicotinamide, and PolypodiumDocument9 pagesDietary Supplements in Dermatology: A Review of The Evidence For Zinc, Biotin, Vitamin D, Nicotinamide, and PolypodiumNazihan Safitri AlkatiriNo ratings yet
- Concept Map MarwahDocument5 pagesConcept Map MarwahAsniah Hadjiadatu AbdullahNo ratings yet
- Herbalage Products KnowledgeDocument25 pagesHerbalage Products Knowledgemail9246No ratings yet
- Outscraper 2024020607591330fd Obstetrician Gynecologist +3Document265 pagesOutscraper 2024020607591330fd Obstetrician Gynecologist +3sanjulata.agriNo ratings yet
- Genomic Characterization of Co-Existing Neoplasia and Carcinoma Lesions Reveals Distinct Evolutionary Paths of Gallbladder CancerDocument11 pagesGenomic Characterization of Co-Existing Neoplasia and Carcinoma Lesions Reveals Distinct Evolutionary Paths of Gallbladder CancerleartaNo ratings yet
- SchizophreniaDocument32 pagesSchizophreniaHerlina SihalohoNo ratings yet
- Reey8t.b. OsteomyelitisDocument52 pagesReey8t.b. Osteomyelitiskuku93No ratings yet
- Improvement of Pregnant Women Visit in Puskesmas As An Effect of Using Ultrasound in Pregnancy ExaminationDocument12 pagesImprovement of Pregnant Women Visit in Puskesmas As An Effect of Using Ultrasound in Pregnancy ExaminationKharinaNo ratings yet
- Hospital Diet and Oral Nutritional Supplements (Sip Feeds)Document16 pagesHospital Diet and Oral Nutritional Supplements (Sip Feeds)Patrick nyawiraNo ratings yet
- 4.3 - Educating Patients, Family, and Health Care Professionals About Foot CareDocument1 page4.3 - Educating Patients, Family, and Health Care Professionals About Foot CareEva Cica SusantiNo ratings yet
- NCMA 219 RLE Laboratory Unit 11bDocument7 pagesNCMA 219 RLE Laboratory Unit 11bJeeduu frostNo ratings yet
- Alcohol Withdrawal - StatPearls - NCBI BookshelfDocument7 pagesAlcohol Withdrawal - StatPearls - NCBI BookshelfDAFNE FRANÇA SANTANANo ratings yet
- In Service Exam For Breast DR Paul BalisiDocument11 pagesIn Service Exam For Breast DR Paul BalisiAmiel Francisco ReyesNo ratings yet
- Nisebj 21 3 2021Document41 pagesNisebj 21 3 2021Clement BewajiNo ratings yet
- Early and Late Preeclamsia Are Characterized by High Cardiac Output, But in The Presence of Fetal Growth Restriction, Cardiac Output Is Low Insights From A Prospective Study PDFDocument12 pagesEarly and Late Preeclamsia Are Characterized by High Cardiac Output, But in The Presence of Fetal Growth Restriction, Cardiac Output Is Low Insights From A Prospective Study PDFJustiawan NazwanNo ratings yet
- Grover 2020Document2 pagesGrover 2020Riza Agung NugrahaNo ratings yet
- BSCPT DU Syllabus-NITOR 4th BatchDocument23 pagesBSCPT DU Syllabus-NITOR 4th BatchHasan Rahman100% (2)
- Blood Vessels QuestionsDocument7 pagesBlood Vessels QuestionsT-Jay Ellis-DaleNo ratings yet
- Pediatric Tonsillectomy PatientsDocument3 pagesPediatric Tonsillectomy PatientsRainer MujicaNo ratings yet