You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (844)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5810)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (347)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Warranty Management and Claims Processing SystemDocument2 pagesWarranty Management and Claims Processing SystemAnkurNo ratings yet
- RCA - WebinarDocument53 pagesRCA - WebinarAnkurNo ratings yet
- Form Master ListDocument1 pageForm Master ListAnkurNo ratings yet
- Effective Process AuditingDocument5 pagesEffective Process AuditingAnkur100% (1)
- Cost of Poor Quality: 1 © Material For QMS COURSE Annexure - 1Document3 pagesCost of Poor Quality: 1 © Material For QMS COURSE Annexure - 1AnkurNo ratings yet
- IATF 16949 Required Documentation For TransitionDocument2 pagesIATF 16949 Required Documentation For TransitionAnkurNo ratings yet
- 1.-Lean WastesDocument22 pages1.-Lean WastesAnkur100% (2)
- QUALITY TALK Apr 2020Document19 pagesQUALITY TALK Apr 2020AnkurNo ratings yet
- Standard Operating Procedure: National Disaster Response ForceDocument20 pagesStandard Operating Procedure: National Disaster Response ForceAnkurNo ratings yet
- HSML COVID-19 Risk Register: MANUFACTURING PLANT: HisarDocument3 pagesHSML COVID-19 Risk Register: MANUFACTURING PLANT: HisarAnkurNo ratings yet
- Application Form For Registration of Assessment AgenciesDocument2 pagesApplication Form For Registration of Assessment AgenciesAnkurNo ratings yet
- Siemens SW Industrial Quality Testing EBDocument12 pagesSiemens SW Industrial Quality Testing EBAnkurNo ratings yet
- Ergonomic Burden CalculatorDocument7 pagesErgonomic Burden CalculatorAnkurNo ratings yet
- Veolia: A Pioneer in The Transition To A Circular Economy: ISWA Annual Congress 2014, Sao Paulo, BrazilDocument20 pagesVeolia: A Pioneer in The Transition To A Circular Economy: ISWA Annual Congress 2014, Sao Paulo, BrazilAnkurNo ratings yet
- End-Of-Line Quality Testing - Automotive Testing International - ArticleDocument1 pageEnd-Of-Line Quality Testing - Automotive Testing International - ArticleAnkurNo ratings yet
- Smart Seat BeltDocument1 pageSmart Seat BeltAnkurNo ratings yet
- Siemens SW Frequently Asked Questions For Transmission Testing With Simcenter Anovis FQDocument5 pagesSiemens SW Frequently Asked Questions For Transmission Testing With Simcenter Anovis FQAnkurNo ratings yet
- Quate For FSSC 22000Document2 pagesQuate For FSSC 22000AnkurNo ratings yet
- Awareness ISO 45001 OHSMSDocument19 pagesAwareness ISO 45001 OHSMSAnkurNo ratings yet
- Course Assignment No - 1Document1 pageCourse Assignment No - 1AnkurNo ratings yet
- Supplier Evaluation For It SMSDocument4 pagesSupplier Evaluation For It SMSAnkurNo ratings yet
- Free Basic Course On Quality Management SystemsDocument2 pagesFree Basic Course On Quality Management SystemsAnkurNo ratings yet
- What Is Quality Function DeploymentDocument6 pagesWhat Is Quality Function DeploymentAnkurNo ratings yet
- Risk Management in Covid Times: USING ISO 31000:2018 Principles in Ealing With A PandemicDocument43 pagesRisk Management in Covid Times: USING ISO 31000:2018 Principles in Ealing With A PandemicAnkurNo ratings yet
- Capabilty Statement Ankur DhirDocument2 pagesCapabilty Statement Ankur DhirAnkurNo ratings yet
- APPRECIATION ONLINE COURSE - MR ShingoDocument3 pagesAPPRECIATION ONLINE COURSE - MR ShingoAnkurNo ratings yet
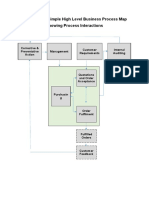
- Example of A Simple High Level Business Process Map Showing Process InteractionsDocument4 pagesExample of A Simple High Level Business Process Map Showing Process InteractionsAnkurNo ratings yet
- Risk Based Thinking ISO 9001-2015Document3 pagesRisk Based Thinking ISO 9001-2015AnkurNo ratings yet