You might also like
- Quality Assurance and Quality Management in Pharmaceutical IndustryFrom EverandQuality Assurance and Quality Management in Pharmaceutical IndustryRating: 4 out of 5 stars4/5 (4)
- A Risk Based Approach To GMP TrainingDocument7 pagesA Risk Based Approach To GMP TrainingRafat AlghubariNo ratings yet
- GMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsFrom EverandGMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsRating: 5 out of 5 stars5/5 (2)
- 014 Quality Unit Roles and ResponsibilitiesDocument35 pages014 Quality Unit Roles and ResponsibilitiesSIRAJ KP100% (1)
- Validation PolicyDocument3 pagesValidation PolicyneppoanandNo ratings yet
- ECA Handling OOE OOT ResultsDocument6 pagesECA Handling OOE OOT Resultsparam540No ratings yet
- EU-GMP.2014 - Annex - 20 (Quality Risk Management)Document25 pagesEU-GMP.2014 - Annex - 20 (Quality Risk Management)bvsc77035No ratings yet
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionFrom EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNo ratings yet
- Nonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsFrom EverandNonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsWilliam J. BrockNo ratings yet
- Current Good Manufacturing Practices (cGMP) for Pharmaceutical ProductsFrom EverandCurrent Good Manufacturing Practices (cGMP) for Pharmaceutical ProductsNo ratings yet
- Practical Approaches to Method Validation and Essential Instrument QualificationFrom EverandPractical Approaches to Method Validation and Essential Instrument QualificationNo ratings yet
- Pharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersFrom EverandPharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersNo ratings yet
- GLP GMP GCPDocument29 pagesGLP GMP GCPmghaffarzadehNo ratings yet
- GMP Class ActivitiesDocument16 pagesGMP Class Activitiescasper_aks100% (1)
- Who Guideline Risk ManagementDocument32 pagesWho Guideline Risk ManagementEka FsNo ratings yet
- Applying ICH Q8 (R2), Q9, and Q10 Principles To CMC ReviewDocument5 pagesApplying ICH Q8 (R2), Q9, and Q10 Principles To CMC Reviewvg_vvgNo ratings yet
- Guide To Inspections of Pharmaceutical Quality Control LaboratoriesDocument16 pagesGuide To Inspections of Pharmaceutical Quality Control Laboratoriessubrata1100% (1)
- Preventing of Cross ContaminationDocument27 pagesPreventing of Cross ContaminationOmar FaruqNo ratings yet
- A Pocket Guide To AuditingDocument10 pagesA Pocket Guide To Auditingmanojdhamne5802No ratings yet
- David L Chesney Parexel International UsaDocument34 pagesDavid L Chesney Parexel International UsaAmer RahmahNo ratings yet
- Continuing Process Verification - 1Document106 pagesContinuing Process Verification - 1Noemi100% (1)
- Compliance Program Program: Chapter 56: Drug Quality AssuranceDocument29 pagesCompliance Program Program: Chapter 56: Drug Quality AssuranceMin Thura OoNo ratings yet
- PICS Quality System RequirementDocument12 pagesPICS Quality System RequirementMd Nasir Uddin KhanNo ratings yet
- Quality Risk Management - The Pharmaceutical Experience PDFDocument25 pagesQuality Risk Management - The Pharmaceutical Experience PDFChiorean Ioana100% (2)
- Contamination Control StrategyDocument13 pagesContamination Control Strategyisrael afolayan mayomiNo ratings yet
- How The Different GLP GMP GCPDocument14 pagesHow The Different GLP GMP GCPawang_timur100% (3)
- Documentation Pharmaceutical IndustryDocument102 pagesDocumentation Pharmaceutical IndustryRagulNo ratings yet
- MHRA GMP Inspection Deficiency Data Trend 2016Document100 pagesMHRA GMP Inspection Deficiency Data Trend 2016Morcos LokaNo ratings yet
- USP 1086 Impurities in Drug Substances and Drug ProductsDocument3 pagesUSP 1086 Impurities in Drug Substances and Drug ProductsMuhammad JamilNo ratings yet
- E06 - Melendez FDA Persp On Multi Product Fac Cross ContaminationDocument22 pagesE06 - Melendez FDA Persp On Multi Product Fac Cross ContaminationppiccoliniNo ratings yet
- FDA - Quality Issues For Clinical Trial MaterialsDocument37 pagesFDA - Quality Issues For Clinical Trial Materialscokekorea100% (1)
- Guidance On Good Data and Record Management PracticesDocument44 pagesGuidance On Good Data and Record Management Practiceskamran alamNo ratings yet
- Pharmaceutical Product Quality Assurance Through CMC Drug Development ProcessDocument20 pagesPharmaceutical Product Quality Assurance Through CMC Drug Development Processpharmashri5399No ratings yet
- PDA 8th Microbiology ConferenceDocument12 pagesPDA 8th Microbiology ConferenceTim SandleNo ratings yet
- Pharmaceutical Quality Control Labs (7 - 93) - FDADocument15 pagesPharmaceutical Quality Control Labs (7 - 93) - FDAdnalokeshNo ratings yet
- Contamination Control Compliance Program PDFDocument7 pagesContamination Control Compliance Program PDFDavid100% (1)
- Aseptic Process ValidationDocument32 pagesAseptic Process ValidationG_Ranjith100% (1)
- Microbial Risk Assesment in Pharmaceutical CleanroomsDocument9 pagesMicrobial Risk Assesment in Pharmaceutical CleanroomsteaNo ratings yet
- Avoiding Errors With The Batch Release ProcessDocument11 pagesAvoiding Errors With The Batch Release ProcessAnthony CollierNo ratings yet
- GMP ManualDocument303 pagesGMP ManualShrinivas TamaskarNo ratings yet
- Validation of HPLC Techniques For Pharmaceutical AnalysisDocument54 pagesValidation of HPLC Techniques For Pharmaceutical Analysisfatos-osmani-5248No ratings yet
- Handling of Deviation: Dr. A. AmsavelDocument34 pagesHandling of Deviation: Dr. A. Amsavelsandro CardosoNo ratings yet
- Common Pharma Interview Questions For FreshersDocument6 pagesCommon Pharma Interview Questions For Freshersrameshwar9595kNo ratings yet
- Guidance For Industry: Process ValidationDocument18 pagesGuidance For Industry: Process ValidationBruno DebonnetNo ratings yet
- A Review On Cleaning Validation in Pharmaceutical IndustryDocument12 pagesA Review On Cleaning Validation in Pharmaceutical IndustryRouag AbdelkarimNo ratings yet
- Pharma Manual PDFDocument25 pagesPharma Manual PDFElena TrofinNo ratings yet
- Pharmaceutical Cleaning A Comprehensive Approach - 0Document15 pagesPharmaceutical Cleaning A Comprehensive Approach - 0Mina Maher MikhailNo ratings yet
- Pharmaceutical Quality Management SystemDocument13 pagesPharmaceutical Quality Management SystemShailesh Gupta100% (1)
- Workshop - Specifications in Early Development (Regulatory Perspective-Stephen Miller, FDA)Document30 pagesWorkshop - Specifications in Early Development (Regulatory Perspective-Stephen Miller, FDA)lhthang1990No ratings yet
- Pharmaceutical Quality Audits: A ReviewDocument9 pagesPharmaceutical Quality Audits: A ReviewHema PepakayalaNo ratings yet
- Drug Shelf LifeDocument27 pagesDrug Shelf LifeAurora SavageNo ratings yet
- Analytical Methods Transfer: Considerations For Biological Products Alfred V. Del Grosso, Ph.D. Fda - CberDocument26 pagesAnalytical Methods Transfer: Considerations For Biological Products Alfred V. Del Grosso, Ph.D. Fda - CberamolNo ratings yet
- QBD Putting Theory Into Practice - Chp2 THE REGULATORY FRAMEWORK PDFDocument26 pagesQBD Putting Theory Into Practice - Chp2 THE REGULATORY FRAMEWORK PDFhdmnauNo ratings yet
- Process Validation of Benazepril HCL 5 MG TabletDocument16 pagesProcess Validation of Benazepril HCL 5 MG TabletCamila Flórez IdárragaNo ratings yet
- Vendor Qualification For Pharmaceutical ExcipientsDocument9 pagesVendor Qualification For Pharmaceutical Excipientsshinta lestari100% (1)
- Impurity ProfileDocument17 pagesImpurity ProfileNishit SuvaNo ratings yet
- Peer Reviewed: Case StudyDocument11 pagesPeer Reviewed: Case StudyPiruzi MaghlakelidzeNo ratings yet
- CGMP GuidelinesDocument23 pagesCGMP GuidelinesRyan 1112No ratings yet
- LAPP KABEL - Wire, Copper Earthing, Transparent, 6 AWG, 16 MM, 4200 X 0.07mm, 164 FT, 50 M - Farnell Element14Document3 pagesLAPP KABEL - Wire, Copper Earthing, Transparent, 6 AWG, 16 MM, 4200 X 0.07mm, 164 FT, 50 M - Farnell Element14two travellerNo ratings yet
- Bending Tools For Press BrakesDocument2 pagesBending Tools For Press BrakespressbraketoolsNo ratings yet
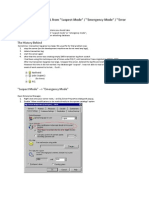
- How To Recover MSSQL From Suspect Mode Emergency Mode Error 1813Document3 pagesHow To Recover MSSQL From Suspect Mode Emergency Mode Error 1813Scott NeubauerNo ratings yet
- Astero E0201E-20 991063Document186 pagesAstero E0201E-20 991063Thinh Nguyen100% (1)
- MGB Torque SettingsDocument3 pagesMGB Torque Settingsdeutscheafrikar100% (1)
- Project Procurement Process ControlDocument4 pagesProject Procurement Process Controlakramsayeed100% (1)
- MIS AssignmentDocument23 pagesMIS AssignmentSharmila PrabhuNo ratings yet
- NTM 2 BaruDocument21 pagesNTM 2 BaruDinie Abdullah ZamawiNo ratings yet
- Gemak Engineering SolutionsDocument22 pagesGemak Engineering Solutionsshashikant.g84No ratings yet
- Aisin Gearbox PartsDocument8 pagesAisin Gearbox Partsthrottleback1812No ratings yet
- ApplicationsdgshteryDocument7 pagesApplicationsdgshterygmik02No ratings yet
- Case Study On An IT (BPO) MNC (Indian)Document6 pagesCase Study On An IT (BPO) MNC (Indian)KNOWLEDGE CREATORSNo ratings yet
- E Biz Spec OverviewDocument50 pagesE Biz Spec OverviewamecobeijingNo ratings yet
- TAQA Approved Contractor and Consultant List 31.05.2021Document342 pagesTAQA Approved Contractor and Consultant List 31.05.2021Bala Krishna100% (1)
- NACTO Urban Bikeway Design GuideDocument29 pagesNACTO Urban Bikeway Design GuideAiman ZhafriNo ratings yet
- Production and Manufacturing Engineering Interview Q and A 1646730474Document6 pagesProduction and Manufacturing Engineering Interview Q and A 1646730474MC ANo ratings yet
- OPM BasicsDocument7 pagesOPM BasicsjeedNo ratings yet
- Procurement Process InnovationDocument23 pagesProcurement Process InnovationakaleembashaNo ratings yet
- Rebar Couplers Brochure 26 01 2017Document32 pagesRebar Couplers Brochure 26 01 2017geraldNo ratings yet
- Maruti Car ManufactureDocument37 pagesMaruti Car ManufactureVema AbhiramNo ratings yet
- Budget 2016-2017 Parks (23 - 05 - 2015)Document187 pagesBudget 2016-2017 Parks (23 - 05 - 2015)Odumodu UmuleriNo ratings yet
- Importance of ISO Certification in Civil EngineeringDocument2 pagesImportance of ISO Certification in Civil EngineeringLuis F. Ruiz GNo ratings yet
- Asset Strategy Development Procedure A2D SAP - 1496221Document11 pagesAsset Strategy Development Procedure A2D SAP - 1496221Alejandro Bartolo YañezNo ratings yet
- CISSP - 9 Buisiness Continuity & Disaster Recovery PlanningDocument53 pagesCISSP - 9 Buisiness Continuity & Disaster Recovery PlanningVeli Anlama100% (4)
- 2001 Ewing Cervero - Travel and Built Environment - A SynthesisDocument28 pages2001 Ewing Cervero - Travel and Built Environment - A SynthesisIrfan Rachman Widjaja K.No ratings yet
- Airvolt TestDocument22 pagesAirvolt Testadam_tomasNo ratings yet
- Farm Jack ManualDocument17 pagesFarm Jack ManualsalahedinNo ratings yet
- MAster Candle FB5 Min 27 SLDocument6 pagesMAster Candle FB5 Min 27 SLRavi VarakalaNo ratings yet
- A395CT 2009 AnswersDocument9 pagesA395CT 2009 AnswersSupuni GunaratneNo ratings yet
- Geto Aluminium Formwork User ManualDocument35 pagesGeto Aluminium Formwork User ManualbsudhareddyNo ratings yet