You might also like
- The Chemistry of Fertilisers and Manure - Including Information on the Chemical Constituents and Types of Fertilisers and ManuresFrom EverandThe Chemistry of Fertilisers and Manure - Including Information on the Chemical Constituents and Types of Fertilisers and ManuresRating: 5 out of 5 stars5/5 (1)
- W W W Rhodium W S Hemistry RchiveDocument2 pagesW W W Rhodium W S Hemistry RchiveMirza CaticNo ratings yet
- 78444Document7 pages78444Silambarasan SivalingamNo ratings yet
- Chemistry Left Over Experiments NarayanaDocument4 pagesChemistry Left Over Experiments Narayananaveenn0803No ratings yet
- Aminative Reduction of KetonesDocument2 pagesAminative Reduction of Ketonesgeovani2100% (1)
- Homogeneous Nonionic Detergent P-N-NonylphenoxydecaethoxyethanolDocument3 pagesHomogeneous Nonionic Detergent P-N-NonylphenoxydecaethoxyethanolMalik FakharNo ratings yet
- 2009 03 05 Glucose AnomersDocument3 pages2009 03 05 Glucose AnomersWouter Nieuwstraten100% (3)
- Glucose Anomers PDFDocument3 pagesGlucose Anomers PDFShahed ANo ratings yet
- Experiment Preparation of P-Nitroacetanilide: StructureDocument3 pagesExperiment Preparation of P-Nitroacetanilide: Structureisma diyanahNo ratings yet
- Synthesis of Novel Fluorobenzothiazole Linked Thiadiazole Compounds: As Possible Anti-Tubercular AgentsDocument2 pagesSynthesis of Novel Fluorobenzothiazole Linked Thiadiazole Compounds: As Possible Anti-Tubercular AgentsVINODNo ratings yet
- Acrylol SynthesisDocument2 pagesAcrylol Synthesis8612106535No ratings yet
- Communications TO EditorDocument2 pagesCommunications TO EditorDPYu1999No ratings yet
- Sternbach Et Al 2002 Quinazolines and 1 4 Benzodiazepines 64 Comparison of The Stereochemistry of Diazepam With That ofDocument4 pagesSternbach Et Al 2002 Quinazolines and 1 4 Benzodiazepines 64 Comparison of The Stereochemistry of Diazepam With That ofNoimurNo ratings yet
- Synthesis of DibenzalacetoneEEEEDocument23 pagesSynthesis of DibenzalacetoneEEEEkylieNo ratings yet
- The Preparation of Pure N-Acetyl-L-leucine and L-Leucine'Document2 pagesThe Preparation of Pure N-Acetyl-L-leucine and L-Leucine'Tuyết MaiNo ratings yet
- Anito SchultzccDocument3 pagesAnito SchultzccJohn PerryNo ratings yet
- PDF Posible Práctica SíntesisDocument7 pagesPDF Posible Práctica SíntesisMarcela MolanoNo ratings yet
- Amilet LucigeninDocument2 pagesAmilet LucigeninKhanh NguyenNo ratings yet
- Carbonyl in Aldehyde & KetoneDocument2 pagesCarbonyl in Aldehyde & KetonetruckerpunkNo ratings yet
- Orbencarb: Materials To Be Analyzed InstrumentationDocument7 pagesOrbencarb: Materials To Be Analyzed Instrumentationjuanvi.sanchoNo ratings yet
- A New Synthesis of MescalineDocument2 pagesA New Synthesis of Mescalinegeovani2No ratings yet
- XylitolDocument3 pagesXylitolTiago Luiz TortellaNo ratings yet
- Notes (CK), Koz-) : of 1,2-Dibrornoacenaphthylene-5,6-Sultone Acenaphthylene-5,6-SultoneDocument2 pagesNotes (CK), Koz-) : of 1,2-Dibrornoacenaphthylene-5,6-Sultone Acenaphthylene-5,6-SultonetrungNo ratings yet
- How To Make DibenzldihydeDocument7 pagesHow To Make DibenzldihydeNirupam GuptaNo ratings yet
- Gluco ZaDocument2 pagesGluco ZaCosmina GeorgianaNo ratings yet
- 39. Dl-β-Phenylalanine - Gillespie - Organic Syntheses Collective 2 (1943) and Organic Syntheses 19 (1939)Document5 pages39. Dl-β-Phenylalanine - Gillespie - Organic Syntheses Collective 2 (1943) and Organic Syntheses 19 (1939)dextroenantiomer100% (1)
- New Synthesis of DiazepamDocument7 pagesNew Synthesis of DiazepamAntônio Neto MachadoNo ratings yet
- Notes 1737: A 100-g. Sample of The Nitro Compound in 100 CCDocument2 pagesNotes 1737: A 100-g. Sample of The Nitro Compound in 100 CCeducobainNo ratings yet
- CHEM 232 - Experiment No. 1 Spring 2018-2019Document2 pagesCHEM 232 - Experiment No. 1 Spring 2018-2019Mohyeddine K. El SayedNo ratings yet
- HydroethylcelluloseDocument6 pagesHydroethylcelluloseAmanah WatiiNo ratings yet
- Experiment 9 PhenacetinDocument2 pagesExperiment 9 PhenacetinShubhNo ratings yet
- Bennett, Pritchard, and Simonsen-: Di.l.zitrodibenza?throneDocument3 pagesBennett, Pritchard, and Simonsen-: Di.l.zitrodibenza?throneAnonymous FigYuONxuuNo ratings yet
- Synthesis of 1,1 - (Butoxymethylene) Dibenzene: Mohamed A. AbukarDocument2 pagesSynthesis of 1,1 - (Butoxymethylene) Dibenzene: Mohamed A. Abukarmohamed abukarNo ratings yet
- Ochem Lab Research PaperDocument2 pagesOchem Lab Research Papermohamed abukarNo ratings yet
- Nuclear Magnetic Resonance Studyes of Triazoles I - Tautomerism of 1,2,4-Triazole - J Org Chem 33 (7), 2956-2957 (1968)Document2 pagesNuclear Magnetic Resonance Studyes of Triazoles I - Tautomerism of 1,2,4-Triazole - J Org Chem 33 (7), 2956-2957 (1968)faustoNo ratings yet
- Suryadatta National School: A.Y. 2022-23 Standard Xii Chemistry Journal IndexDocument7 pagesSuryadatta National School: A.Y. 2022-23 Standard Xii Chemistry Journal IndexKairav Bharat PathakNo ratings yet
- Synthesis of Ketamine PDFDocument4 pagesSynthesis of Ketamine PDFJohn Haas100% (3)
- 3,4-Methylenedioxyphenyl-2-Nitropropene From Piperonal & NitroethaneDocument1 page3,4-Methylenedioxyphenyl-2-Nitropropene From Piperonal & NitroethaneJi ChemNo ratings yet
- Chapter - 2 Synthesis and Purification of Monoazo Disperse WesDocument9 pagesChapter - 2 Synthesis and Purification of Monoazo Disperse WesWalid Ebid ElgammalNo ratings yet
- Ffee 430000004Document2 pagesFfee 430000004Anonymous FigYuONxuuNo ratings yet
- Experiment 1: Spectroscopic and Magnetochemical Properties of Ni (II) Complexes in Different GeometriesDocument4 pagesExperiment 1: Spectroscopic and Magnetochemical Properties of Ni (II) Complexes in Different GeometriesMohyeddine K. El SayedNo ratings yet
- Preperatory TechniquesDocument24 pagesPreperatory TechniquessundaasNo ratings yet
- Histamine PhosphateDocument1 pageHistamine Phosphategrace_febiantyNo ratings yet
- Amsterdamsky J. Chem. Ed. 1998Document2 pagesAmsterdamsky J. Chem. Ed. 1998Ahmed. bdbNo ratings yet
- Medicinal Chemistry 8th Semester PracticalDocument11 pagesMedicinal Chemistry 8th Semester PracticalMohammad Nasim AkhtarNo ratings yet
- Section-B: A Practical Book of Medicinal Chemistry-I (Sem. - V)Document19 pagesSection-B: A Practical Book of Medicinal Chemistry-I (Sem. - V)Raju NiraulaNo ratings yet
- Inorganic ChemistryDocument5 pagesInorganic ChemistryKatherine AlvarezNo ratings yet
- Synthesis of Acetophenone DerivativesDocument6 pagesSynthesis of Acetophenone DerivativesAwad SaidNo ratings yet
- Lah Nitroalkenes - Reverse AdditionDocument7 pagesLah Nitroalkenes - Reverse AdditionGindo Baroes WanNo ratings yet
- The Chemistry Dicyclopentadiene. Hydration and RearrangementDocument6 pagesThe Chemistry Dicyclopentadiene. Hydration and Rearrangementmanuel querolNo ratings yet
- Preparation of n-Isopropylidene-n'-2-Nitrobenzenesulfonyl Hydrazine (IPNBSH) and Its Use in Palladium-Catalyzed Synthesis of Monoalkyl Diazenes. Synthesis of 9-AllylanthraceneDocument22 pagesPreparation of n-Isopropylidene-n'-2-Nitrobenzenesulfonyl Hydrazine (IPNBSH) and Its Use in Palladium-Catalyzed Synthesis of Monoalkyl Diazenes. Synthesis of 9-AllylanthraceneYami BaudelaireNo ratings yet
- Chemistry Unit 1Document16 pagesChemistry Unit 1Bhairavi MNo ratings yet
- The Gabriel Synthesis of Benzylamine: An Undergraduate Organic ExperimentDocument2 pagesThe Gabriel Synthesis of Benzylamine: An Undergraduate Organic ExperimentJuan MartínezNo ratings yet
- DexpanthenolDocument2 pagesDexpanthenolArtem KulikovNo ratings yet
- Bergel and Morrison T H e Oxidation of Adrenaline. AdrenalineDocument1 pageBergel and Morrison T H e Oxidation of Adrenaline. AdrenalineAnonymous FigYuONxuuNo ratings yet
- A New Technique in Preparing 2,4-Dinitrophenylhydrazones.Document2 pagesA New Technique in Preparing 2,4-Dinitrophenylhydrazones.H Vásquez GalindoNo ratings yet
- Bergel and Morrison 5-Hydroxyindole. 5-Hydroxyindole.: 5-Methoxyindole-2-Carboxylic 2-Nitro-5-HydroxytolueneDocument1 pageBergel and Morrison 5-Hydroxyindole. 5-Hydroxyindole.: 5-Methoxyindole-2-Carboxylic 2-Nitro-5-HydroxytolueneAnonymous FigYuONxuuNo ratings yet
- Ethene Homo - and Ethenepropene Copolymerization 2000Document5 pagesEthene Homo - and Ethenepropene Copolymerization 2000mayamaruguerraNo ratings yet
- Synthesis of NitrazepamDocument3 pagesSynthesis of Nitrazepamjiskate77100% (1)
- Exp 4 - P-Nitro AcetanilideDocument3 pagesExp 4 - P-Nitro AcetanilideDeep Dave50% (2)
- Pa 09 02050Document7 pagesPa 09 02050Luciano PaoloNo ratings yet
- Certificado Imp C Ibuprofeno USP - 1335541-R06930Document3 pagesCertificado Imp C Ibuprofeno USP - 1335541-R06930Luciano PaoloNo ratings yet
- 1 s2.0 S004040391830039X MainDocument4 pages1 s2.0 S004040391830039X MainLuciano PaoloNo ratings yet
- Anal Bioanal Chem (2015) 407,3115-3123Document9 pagesAnal Bioanal Chem (2015) 407,3115-3123Luciano PaoloNo ratings yet
- 03639045.2020.1821054 Metodo HPLC Paracetamol Imp JDocument29 pages03639045.2020.1821054 Metodo HPLC Paracetamol Imp JLuciano PaoloNo ratings yet
- SDS Ibuprofeno Imp CDocument8 pagesSDS Ibuprofeno Imp CLuciano PaoloNo ratings yet
- Joc 1987 + KohDocument5 pagesJoc 1987 + KohLuciano PaoloNo ratings yet
- Metrological Traceability Chain For PCBs I.N.Ri.M. ActivityDocument5 pagesMetrological Traceability Chain For PCBs I.N.Ri.M. ActivityLuciano PaoloNo ratings yet
- bmr043 Metodo HPLC Paracetamol Imp JDocument8 pagesbmr043 Metodo HPLC Paracetamol Imp JLuciano PaoloNo ratings yet
- Development Studies of A New Metronidazole CertifiDocument11 pagesDevelopment Studies of A New Metronidazole CertifiLuciano PaoloNo ratings yet
- Estimación de PM Using PGFsDocument4 pagesEstimación de PM Using PGFsLuciano PaoloNo ratings yet
- Importance of Purity Evaluation and The Potential of Quantitative 1H NMR As A Purity AssayDocument12 pagesImportance of Purity Evaluation and The Potential of Quantitative 1H NMR As A Purity AssayLuciano PaoloNo ratings yet
- CMR A 21422Document8 pagesCMR A 21422Luciano PaoloNo ratings yet
- Chemcomm: CommunicationDocument4 pagesChemcomm: CommunicationLuciano PaoloNo ratings yet
- Jacs (2008) - 130 - 10521-10523 DafDocument3 pagesJacs (2008) - 130 - 10521-10523 DafLuciano PaoloNo ratings yet
- New J. Chem. (2019), 43,14028-14035Document15 pagesNew J. Chem. (2019), 43,14028-14035Luciano PaoloNo ratings yet
- Molecules (2020) - 25 - 3793-3803 DADocument10 pagesMolecules (2020) - 25 - 3793-3803 DALuciano PaoloNo ratings yet
- Anal. Chem. 2018 NIST PS1 Primary NMR Standar (Benzoic Acid)Document8 pagesAnal. Chem. 2018 NIST PS1 Primary NMR Standar (Benzoic Acid)Luciano PaoloNo ratings yet
- NMR Spectroscopy For Metabolomics and Metabolic Pro FilingDocument14 pagesNMR Spectroscopy For Metabolomics and Metabolic Pro FilingLuciano PaoloNo ratings yet
- Tetrehedron (2011) - 67 - 3464-3469 Ref 21 de Molecules 2020Document6 pagesTetrehedron (2011) - 67 - 3464-3469 Ref 21 de Molecules 2020Luciano PaoloNo ratings yet
- J Fluorine Chem 113 (2002) 65-78Document14 pagesJ Fluorine Chem 113 (2002) 65-78Luciano PaoloNo ratings yet
- 1 s2.0 S0968089612004567 MainDocument8 pages1 s2.0 S0968089612004567 MainLuciano PaoloNo ratings yet
- J Fluorine Chem 106 (2000) 43-52Document10 pagesJ Fluorine Chem 106 (2000) 43-52Luciano PaoloNo ratings yet
- Mccombie, Stacey: SaundersDocument3 pagesMccombie, Stacey: SaundersLuciano PaoloNo ratings yet
- J Fluorine Chem 106 (2000) 153-161Document9 pagesJ Fluorine Chem 106 (2000) 153-161Luciano PaoloNo ratings yet
- Russ Chem Bull - 1999 - 48 - 2 - 367Document4 pagesRuss Chem Bull - 1999 - 48 - 2 - 367Luciano PaoloNo ratings yet
- Fmolb 09 882487Document12 pagesFmolb 09 882487Luciano PaoloNo ratings yet
- Jo200522w Si 002Document46 pagesJo200522w Si 002Luciano PaoloNo ratings yet
- Phosphorus, Sulfur, and Silicon and The Related Elements - 2010 - 185, 11, 2182Document14 pagesPhosphorus, Sulfur, and Silicon and The Related Elements - 2010 - 185, 11, 2182Luciano PaoloNo ratings yet
- INMETRO DicloDocument12 pagesINMETRO DicloLuciano PaoloNo ratings yet
- Wastewater TreatmentDocument61 pagesWastewater TreatmentGoutham R100% (1)
- Past Questions According To Article: Multiple Choice Questions LiquidsDocument8 pagesPast Questions According To Article: Multiple Choice Questions LiquidsSheikh Shahbaz AliNo ratings yet
- IndexDocument17 pagesIndexAmarendra TripathyNo ratings yet
- Lecture 1-Atoms, Molecules & IonsDocument18 pagesLecture 1-Atoms, Molecules & Ionslim_zeyongNo ratings yet
- Pembentukan Dan Karakterisasi Dispersi Padat Efavirenzs-CrospovidoneDocument7 pagesPembentukan Dan Karakterisasi Dispersi Padat Efavirenzs-CrospovidoneanggiberNo ratings yet
- Tutorial Chapter 1 - 0 ThermodynamicDocument2 pagesTutorial Chapter 1 - 0 ThermodynamicSufferedMuchNo ratings yet
- AEMTC LPG Liquid Cargo Handling Simulator Course Handout A 3 Day Course For OfficersDocument89 pagesAEMTC LPG Liquid Cargo Handling Simulator Course Handout A 3 Day Course For OfficersPhan DungNo ratings yet
- PFR Data AnalysisDocument46 pagesPFR Data AnalysisAkk KolNo ratings yet
- Distillation Lecture Note-2Document20 pagesDistillation Lecture Note-2BasseyNo ratings yet
- Energy Balance of SolutionsDocument16 pagesEnergy Balance of Solutionsnhalieza1067No ratings yet
- DEHYDRATION OF p-MENTHAN-3-OL (MENTHOL)Document5 pagesDEHYDRATION OF p-MENTHAN-3-OL (MENTHOL)winfredNo ratings yet
- The Nature of Reactants Is Different From The OtheDocument1 pageThe Nature of Reactants Is Different From The OthelisaNo ratings yet
- Synthesis of Some New Substituted 1,2,4-Triazole and 1,3,4-Thiadiazole and Study of Their Activities On Some Strains of BacteriaDocument8 pagesSynthesis of Some New Substituted 1,2,4-Triazole and 1,3,4-Thiadiazole and Study of Their Activities On Some Strains of BacteriaAmer KasidehNo ratings yet
- AssignmentDocument2 pagesAssignmentMark Lester RealNo ratings yet
- Glass-Ceramics For Photonic Applications: An ExampleDocument22 pagesGlass-Ceramics For Photonic Applications: An ExamplemadangkNo ratings yet
- Assignment 1 RACDocument2 pagesAssignment 1 RACShridhar Walavalkar50% (2)
- 1 s2.0 S0026265X22011298 MainDocument7 pages1 s2.0 S0026265X22011298 MainMohammad Imran HossainNo ratings yet
- Chapter 20 PolymerDocument24 pagesChapter 20 PolymerVidya PasalkarNo ratings yet
- Light Propagation in Optical FiberDocument22 pagesLight Propagation in Optical FiberBhagyalaxmi BeheraNo ratings yet
- Electron ConfigurationDocument28 pagesElectron ConfigurationEbb Edel QuibodNo ratings yet
- Investigatory Project 2019Document10 pagesInvestigatory Project 2019Drexel Cueto0% (2)
- Poly (Vinylidene Fluoride) Clay Nanocomposites Prepared byDocument8 pagesPoly (Vinylidene Fluoride) Clay Nanocomposites Prepared byPrapto PraptNo ratings yet
- 11 PsychrometricsDocument13 pages11 PsychrometricsImranAtheeqNo ratings yet
- Fuderer, A. and Rudelstorfer, E. Selective Adsorption Process. U.S. Patent 3,986,849Document27 pagesFuderer, A. and Rudelstorfer, E. Selective Adsorption Process. U.S. Patent 3,986,849kay LauNo ratings yet
- PH3256 DHDocument19 pagesPH3256 DHdigitalepcsNo ratings yet
- TB ch03Document11 pagesTB ch03Rica RoscoNo ratings yet
- Assignment 2Document2 pagesAssignment 2vijendra mauryaNo ratings yet
- Semester-IV Chemistry Paper-V Syllabus and Model PaperDocument5 pagesSemester-IV Chemistry Paper-V Syllabus and Model PaperVamsi ArisettiNo ratings yet
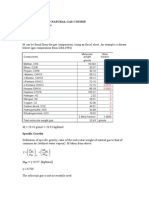
- Calculations in Natural Gas CourseDocument6 pagesCalculations in Natural Gas Coursepatrickandreas77No ratings yet
- Lloyd S Register Energy Guidance Notes For Risk Based Ana PDFDocument22 pagesLloyd S Register Energy Guidance Notes For Risk Based Ana PDFankitalalwaniNo ratings yet
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (5)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincFrom EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincRating: 3.5 out of 5 stars3.5/5 (137)
- AP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeFrom EverandAP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeRating: 5 out of 5 stars5/5 (1)
- It's Elemental: The Hidden Chemistry in EverythingFrom EverandIt's Elemental: The Hidden Chemistry in EverythingRating: 4 out of 5 stars4/5 (10)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeFrom EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeRating: 5 out of 5 stars5/5 (4)
- Taste: Surprising Stories and Science About Why Food Tastes GoodFrom EverandTaste: Surprising Stories and Science About Why Food Tastes GoodRating: 3 out of 5 stars3/5 (20)
- Guidelines for Defining Process Safety Competency RequirementsFrom EverandGuidelines for Defining Process Safety Competency RequirementsRating: 3 out of 5 stars3/5 (1)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeFrom EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeRating: 4 out of 5 stars4/5 (1)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (14)
- Formulating, Packaging, and Marketing of Natural Cosmetic ProductsFrom EverandFormulating, Packaging, and Marketing of Natural Cosmetic ProductsNo ratings yet
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideFrom EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideNo ratings yet
- The Periodic Table: A Very Short IntroductionFrom EverandThe Periodic Table: A Very Short IntroductionRating: 4.5 out of 5 stars4.5/5 (3)
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeFrom EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNo ratings yet
- The Billion-Dollar Molecule: The Quest for the Perfect DrugFrom EverandThe Billion-Dollar Molecule: The Quest for the Perfect DrugRating: 5 out of 5 stars5/5 (2)
- Guidelines for Integrating Process Safety into Engineering ProjectsFrom EverandGuidelines for Integrating Process Safety into Engineering ProjectsNo ratings yet
- Fundamentals of Chemistry: A Modern IntroductionFrom EverandFundamentals of Chemistry: A Modern IntroductionRating: 5 out of 5 stars5/5 (1)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (90)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (1)
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolFrom EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNo ratings yet
- Tribology: Friction and Wear of Engineering MaterialsFrom EverandTribology: Friction and Wear of Engineering MaterialsRating: 5 out of 5 stars5/5 (1)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsFrom EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsRating: 4 out of 5 stars4/5 (146)