You might also like
- AntibioticsDocument53 pagesAntibioticsMaheen IdreesNo ratings yet
- Cell Wall Inhibitor PPT SlideDocument47 pagesCell Wall Inhibitor PPT Slidekhawaja sahabNo ratings yet
- AntibioticsDocument6 pagesAntibioticsManzoor AhmadNo ratings yet
- Antibiotics CologyDocument31 pagesAntibiotics CologyManthan ChauhanNo ratings yet
- PenicillinDocument21 pagesPenicillinnadar shahNo ratings yet
- CLINICAL USE OF ANTIBIOTICS IN VETERINARY PRACTICEDocument83 pagesCLINICAL USE OF ANTIBIOTICS IN VETERINARY PRACTICEhansmeet100% (1)
- STREPTOMYCINDocument3 pagesSTREPTOMYCINChad InongNo ratings yet
- Penicillins: Classification, Mechanism of Action & UsesDocument58 pagesPenicillins: Classification, Mechanism of Action & UsesSally PujaNo ratings yet
- Cell wall synthesis inhibitors: Beta-Lactam antibioticsDocument48 pagesCell wall synthesis inhibitors: Beta-Lactam antibioticsmulatumeleseNo ratings yet
- Essential Guide to PenicillinsDocument8 pagesEssential Guide to Penicillinsgulshan araNo ratings yet
- Antibiotics Simplified 1st Edition DR - Osama Ma3rofDocument57 pagesAntibiotics Simplified 1st Edition DR - Osama Ma3rofDark AngelNo ratings yet
- CHEMOTHERAPY FOR TUBERCULOSISDocument49 pagesCHEMOTHERAPY FOR TUBERCULOSISJia YingNo ratings yet
- AntibioticsDocument51 pagesAntibioticsClarenceNo ratings yet
- Antimicrobial Drugs: Powerpoint Presentations Prepared by Bradley W. Christian, Mclennan Community CollegeDocument35 pagesAntimicrobial Drugs: Powerpoint Presentations Prepared by Bradley W. Christian, Mclennan Community CollegeTiffany Jane Huertas100% (1)
- Romeo Victor M. Valderrama BSN-2A: CNS: Confusion, Depression, BeforeDocument8 pagesRomeo Victor M. Valderrama BSN-2A: CNS: Confusion, Depression, BeforeitsmeayaNo ratings yet
- Antibiotics: Classification and ExamplesDocument39 pagesAntibiotics: Classification and ExamplesV G Viju KumarNo ratings yet
- PENICILLINSDocument109 pagesPENICILLINSAnamta AshfaqNo ratings yet
- Antibiotics Classification and MechanismsDocument20 pagesAntibiotics Classification and MechanismsSalmanNo ratings yet
- AntibioticsDocument18 pagesAntibioticsAhnaf Muttaqi Spondan 1716No ratings yet
- Beta-Lactam Antibiotics & Other Inhibitors of Cell WallDocument71 pagesBeta-Lactam Antibiotics & Other Inhibitors of Cell WallAlvin LaurenceNo ratings yet
- Beta Lactams 1Document44 pagesBeta Lactams 1C E princeyNo ratings yet
- Cell Wall Inhibitors: Zarqa University Pharmacy School Clinical Pharmacy and Therapeutics Department Pharmacology IIIDocument45 pagesCell Wall Inhibitors: Zarqa University Pharmacy School Clinical Pharmacy and Therapeutics Department Pharmacology IIIAla'a AL AqrabawyNo ratings yet
- Lecture 7 Chemotherapeutic AgentDocument54 pagesLecture 7 Chemotherapeutic AgentjowditzNo ratings yet
- Uc PDFDocument19 pagesUc PDFPenNo ratings yet
- Antibiotics Classification GuideDocument16 pagesAntibiotics Classification GuideFarida CitraNo ratings yet
- PenicillinDocument3 pagesPenicillinIremey ReyesNo ratings yet
- PenicillinsDocument15 pagesPenicillinsFaisalNo ratings yet
- Beta Lactum AntibioticsDocument26 pagesBeta Lactum AntibioticsEmon KhanNo ratings yet
- PenicilinasDocument19 pagesPenicilinasVALENTINA ZAMBRANO ROMERONo ratings yet
- Antibiotic 1-Terminology: 1. Inhibition of Cell Wall SynthesisDocument12 pagesAntibiotic 1-Terminology: 1. Inhibition of Cell Wall SynthesismidoNo ratings yet
- The Penicillins: Symposium On Antimicrobial Agents-Part VIDocument18 pagesThe Penicillins: Symposium On Antimicrobial Agents-Part VIAdrián LovonyákNo ratings yet
- Penicillin ProductionDocument11 pagesPenicillin ProductionPragna BellaNo ratings yet
- Industrial Production of PenicillinDocument9 pagesIndustrial Production of Penicillinssthakurleo100% (11)
- Cell Wall Inhibitors Target Penicillin-Binding ProteinsDocument61 pagesCell Wall Inhibitors Target Penicillin-Binding ProteinsFawaazulla KhanNo ratings yet
- What Antibiotics Are: Sources, Mechanisms and ClassificationDocument51 pagesWhat Antibiotics Are: Sources, Mechanisms and ClassificationAnnie KhanNo ratings yet
- Mechanism of Action of Penicillin & CephalosporinDocument7 pagesMechanism of Action of Penicillin & CephalosporinshahabahmadNo ratings yet
- Penicillin and Semisynthetic Penicillins in Dermatology: Disease-a-Month June 2004Document13 pagesPenicillin and Semisynthetic Penicillins in Dermatology: Disease-a-Month June 2004Darajjee mokonninNo ratings yet
- Cell Wall Inhibitors: Beta-Lactams: PenicillinsDocument12 pagesCell Wall Inhibitors: Beta-Lactams: Penicillinsمحمد بن صالحNo ratings yet
- Faculty: Dr. Alvin Fox: Suggested Reading: Murray, Third Edition Chapters 10 & 20Document23 pagesFaculty: Dr. Alvin Fox: Suggested Reading: Murray, Third Edition Chapters 10 & 20darshan20022No ratings yet
- Abiola Muhammad, ADEOSUN Department of Biochemistry, Lead City University, IbadanDocument37 pagesAbiola Muhammad, ADEOSUN Department of Biochemistry, Lead City University, IbadanKhushi PatelNo ratings yet
- Unit 1 - MC 3Document58 pagesUnit 1 - MC 3Shreyas ShreyuNo ratings yet
- Beta-Lactam & Other Cell Wall-& Membrane-Active AntibioticsDocument43 pagesBeta-Lactam & Other Cell Wall-& Membrane-Active AntibioticsTutde SedanaNo ratings yet
- (Lec-8) Cell Wall Synthesis, 2-10-2023Document46 pages(Lec-8) Cell Wall Synthesis, 2-10-2023Muhammad Zahid Muhammad ZahidNo ratings yet
- Antimicrobial Drugs: Laboratory of Microbiology Medical Faculty Brawijaya UniversityDocument38 pagesAntimicrobial Drugs: Laboratory of Microbiology Medical Faculty Brawijaya UniversityYuu Ayu'k LifestarNo ratings yet
- PenicillinsDocument7 pagesPenicillinsZain BaderNo ratings yet
- Antibiotics Targeting Cell Wall Synthesis in BacteriaDocument4 pagesAntibiotics Targeting Cell Wall Synthesis in BacteriaMuhammad NawazNo ratings yet
- Chapter 10. Antibiotics and Antimicrobial TestDocument76 pagesChapter 10. Antibiotics and Antimicrobial TestThành NamNo ratings yet
- AntibioticsDocument168 pagesAntibioticsamirabadr2005No ratings yet
- Antibacterial Drugs Cellwall and Protein Synthesis InhihibitorsDocument41 pagesAntibacterial Drugs Cellwall and Protein Synthesis InhihibitorsWezzyNo ratings yet
- Chapter 43 - Beta-LactamDocument7 pagesChapter 43 - Beta-LactamErika De JesusNo ratings yet
- PenicillinDocument14 pagesPenicillinVeena PatilNo ratings yet
- Antibacterial AntibioticsDocument13 pagesAntibacterial AntibioticsMuhamed ArsalanNo ratings yet
- Antimicrobials Revision: Gram Staining, MIC, Antibiotic Misuse & MechanismsDocument5 pagesAntimicrobials Revision: Gram Staining, MIC, Antibiotic Misuse & MechanismsDanny LeeNo ratings yet
- Antibiotic Resistance PDFDocument6 pagesAntibiotic Resistance PDFGea MarieNo ratings yet
- 2-Cell Wall Inhibitor Antibitics-I Oct 15th 2018Document28 pages2-Cell Wall Inhibitor Antibitics-I Oct 15th 2018Leena AlateeqNo ratings yet
- ANTIBIOTICS IN ORAL SURGERYs 123Document43 pagesANTIBIOTICS IN ORAL SURGERYs 123Puneet SinghNo ratings yet
- Shakil 2007Document10 pagesShakil 2007Leila RaNo ratings yet
- Phar - Cell Wall Inhibitor - Lec 7 - TheoDocument20 pagesPhar - Cell Wall Inhibitor - Lec 7 - Theoسلام شاكر حميد جميل 6506No ratings yet
- AntibioticsDocument5 pagesAntibioticsSneeha VeerakumarNo ratings yet
- Week 4 - Penicillins, CephalosporinsDocument44 pagesWeek 4 - Penicillins, CephalosporinsTiko JomidavaNo ratings yet
- penicillin-140625060421-phpapp01-convertedDocument22 pagespenicillin-140625060421-phpapp01-convertednewtamil 2021No ratings yet
- 7610 (24) Antimicrobial Drugs I (Antibiotics) - UpdatedDocument59 pages7610 (24) Antimicrobial Drugs I (Antibiotics) - UpdatedAli AlhayaliNo ratings yet
- Lecture 11 - 306-Antimicrobial - QDocument19 pagesLecture 11 - 306-Antimicrobial - QFaysal HassanNo ratings yet
- Antibiotics: April 2016Document25 pagesAntibiotics: April 2016Santiago LopezNo ratings yet
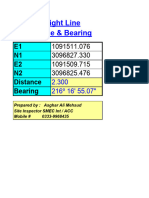
- 8 .Distance BearingDocument3 pages8 .Distance BearingSyed Ali ShahNo ratings yet
- TraverseXL V1 0Document61 pagesTraverseXL V1 0SyarifNo ratings yet
- Typical Crass Section 0001Document8 pagesTypical Crass Section 0001Syed Ali ShahNo ratings yet
- Typical Crass Section 0001Document8 pagesTypical Crass Section 0001Syed Ali ShahNo ratings yet
- Passport Apply FormDocument1 pagePassport Apply FormSyed Ali ShahNo ratings yet
- Pesco Online BillDocument2 pagesPesco Online BillSyed Ali ShahNo ratings yet
- PESCO ONLINE BILL NovemberDocument2 pagesPESCO ONLINE BILL NovemberSyed Ali ShahNo ratings yet
- PESCO ONLINE BILL December 2022Document2 pagesPESCO ONLINE BILL December 2022Syed Ali ShahNo ratings yet
- k12 Curriculum Guides Science Grade8 Unit 4Document30 pagesk12 Curriculum Guides Science Grade8 Unit 4Syed Ali ShahNo ratings yet
- Exam 3 Pharm QuestionsDocument10 pagesExam 3 Pharm QuestionsUneke EdwardsNo ratings yet
- LAHORE MEDICAL COLLEGE CHEMOTHERAPY EXAMDocument36 pagesLAHORE MEDICAL COLLEGE CHEMOTHERAPY EXAMClinicalTraining0% (1)
- Abdi Et Al. 2008Document6 pagesAbdi Et Al. 2008argos1301No ratings yet
- Research Proposal Outline for Studying Effects of Diclofenac on ZebrafishDocument44 pagesResearch Proposal Outline for Studying Effects of Diclofenac on ZebrafishJolaine ValloNo ratings yet
- Drug Development Against Tuberculosis Impact of AlkaloidsDocument41 pagesDrug Development Against Tuberculosis Impact of Alkaloidsamine ben dahsenNo ratings yet
- Acupuncture ItrDocument85 pagesAcupuncture ItrVpr OmNo ratings yet
- Meier Et Al, 1996Document4 pagesMeier Et Al, 1996boni_sebayangNo ratings yet
- Streptomycetes Research ArticleDocument12 pagesStreptomycetes Research Articleapi-637941041No ratings yet
- Allergic Contact Dermatitis To Topical Antibiotics: Epidemiology, Responsible Allergens, and ManagementDocument21 pagesAllergic Contact Dermatitis To Topical Antibiotics: Epidemiology, Responsible Allergens, and ManagementCrhistian Toribio DionicioNo ratings yet
- MCB 308 Pharmaceutical Microbiology PDFDocument23 pagesMCB 308 Pharmaceutical Microbiology PDFIlori OluwafemiNo ratings yet
- Determination of Aminoglycoside Antibiotics, Current Status andDocument19 pagesDetermination of Aminoglycoside Antibiotics, Current Status andMuhammad SajjadNo ratings yet
- Anti TB DrugsDocument23 pagesAnti TB DrugsAbiHa YousaufNo ratings yet
- Antimicrobial Drugs - Neeraj DhillonDocument16 pagesAntimicrobial Drugs - Neeraj DhillonNeeraj DhillonNo ratings yet
- Buruli Ulcer PDFDocument73 pagesBuruli Ulcer PDFMaria Del Mar RoblesNo ratings yet
- Ch12 Part1Document15 pagesCh12 Part1lasanders601No ratings yet
- Aminoglycoside Antibiotics: Mechanism of ActionDocument9 pagesAminoglycoside Antibiotics: Mechanism of Actionprabhakaran payamNo ratings yet
- AMINOGLYCOSIDESDocument15 pagesAMINOGLYCOSIDESGareth BaleNo ratings yet
- Pmoc Practice Test 300 QSDocument12 pagesPmoc Practice Test 300 QSJohn MelbyNo ratings yet
- Drug Study QiDocument6 pagesDrug Study QiKevin Sam AguirreNo ratings yet
- Antimicrobial DrugsDocument24 pagesAntimicrobial DrugsMuh Akbar BaharNo ratings yet
- Dosage: Route:: Mycobacterium TuberculosisDocument21 pagesDosage: Route:: Mycobacterium TuberculosisLyn ConsingNo ratings yet
- Aminoglycoside Antibiotics and Skeletal Muscle Relaxants: SourcesDocument2 pagesAminoglycoside Antibiotics and Skeletal Muscle Relaxants: SourcesahmedNo ratings yet