You might also like
- GB T700-2006Carbonstructuralsteels (英文版)Document11 pagesGB T700-2006Carbonstructuralsteels (英文版)yong liNo ratings yet
- HW4 ProteinsDocument3 pagesHW4 ProteinsFATIMA AIRA LEGASPINo ratings yet
- Cytec-Solutions-Vol 16 Solvent ExtractionDocument39 pagesCytec-Solutions-Vol 16 Solvent ExtractionAbrahan BVNo ratings yet
- IGCSE Chemistry Paper 6Document18 pagesIGCSE Chemistry Paper 6Imen KsibiNo ratings yet
- Quality Control Routines in A Dairy IndustryDocument75 pagesQuality Control Routines in A Dairy IndustryVũ Thế AnhNo ratings yet
- EXPERIMENT 1 - Reactions of Aliphatic and Aromatic HydrocarbonsDocument2 pagesEXPERIMENT 1 - Reactions of Aliphatic and Aromatic HydrocarbonsASYRANI ZULAIKHANo ratings yet
- Bloembergen-Spin Relaxation Processes in A Two-Proton SystemDocument6 pagesBloembergen-Spin Relaxation Processes in A Two-Proton SystemIsra RMNo ratings yet
- 2022 - JRWang - ArXiv - Controlling Frustrated Magnetism On The Kagome Lattice by Uniaxial-Strain TuningDocument7 pages2022 - JRWang - ArXiv - Controlling Frustrated Magnetism On The Kagome Lattice by Uniaxial-Strain TuningshuoliNo ratings yet
- Electroabsorption and Related Spectroscopic Studies of Bimetallic Tetraiminoethylenedimacrocyclic Complexes: Corroboration of Valence Electron DelocalizationDocument4 pagesElectroabsorption and Related Spectroscopic Studies of Bimetallic Tetraiminoethylenedimacrocyclic Complexes: Corroboration of Valence Electron DelocalizationAzizah MunitaNo ratings yet
- Continentino1983 FerromagnetsDocument6 pagesContinentino1983 FerromagnetsMylena Pinto NascimentoNo ratings yet
- Teixidor 1987Document2 pagesTeixidor 1987agcfilesloverNo ratings yet
- Entropy-Driven Phase TransitionsDocument13 pagesEntropy-Driven Phase TransitionsCat SkullNo ratings yet
- Eaat2374 FullDocument8 pagesEaat2374 FulljdslkfjlAKNo ratings yet
- Time QuantDocument3 pagesTime QuantAlex TernovNo ratings yet
- Can Self-Replicating Species Flourish in The Interior of A StarDocument4 pagesCan Self-Replicating Species Flourish in The Interior of A StarMuhammad Ibrahim AbdulhamidNo ratings yet
- Beirau Et Al. - 2019 - Avalanches During Recrystallization in Radiation-Damaged Pyrochlore and AllaniteDocument5 pagesBeirau Et Al. - 2019 - Avalanches During Recrystallization in Radiation-Damaged Pyrochlore and AllaniteAnna ShelyugNo ratings yet
- What Drives Nematic Order in Iron-Based Superconductors?: Review ArticleDocument8 pagesWhat Drives Nematic Order in Iron-Based Superconductors?: Review Article142520No ratings yet
- Examples of Electrocyclic ReactionsDocument14 pagesExamples of Electrocyclic ReactionsAn TruongNo ratings yet
- Gas Dynamics in Clusters of GalaxiesDocument30 pagesGas Dynamics in Clusters of GalaxiespjblkNo ratings yet
- ElectrocyclicDocument22 pagesElectrocyclicCreative ThinkerNo ratings yet
- Woestenenk NimbDocument10 pagesWoestenenk NimbFundaljohnNo ratings yet
- 2017 Rupan LCO ArxivDocument10 pages2017 Rupan LCO Arxivamol476853No ratings yet
- Lower-Entropy ArticleDocument27 pagesLower-Entropy ArticleCameron GrahamNo ratings yet
- International Journal of Non-Linear Mechanics: Ivana Kovacic, Michael J. Brennan, Benjamin LinetonDocument10 pagesInternational Journal of Non-Linear Mechanics: Ivana Kovacic, Michael J. Brennan, Benjamin LinetonBehzad BehdaniNo ratings yet
- On The Lattice Ground State of Densely Packed Hard EllipsesDocument5 pagesOn The Lattice Ground State of Densely Packed Hard Ellipsesbosheng dingNo ratings yet
- Feynman. The Theory of PositronsDocument11 pagesFeynman. The Theory of PositronsricardosoaresvieiraNo ratings yet
- Carbanionic Rearrangements of Halomethylenecyclobutanes. Stereochemistry of The Migrating GroupDocument12 pagesCarbanionic Rearrangements of Halomethylenecyclobutanes. Stereochemistry of The Migrating GroupDiogo DiasNo ratings yet
- Non-Collinear Magnetism Due To Orbital Degeneracy and Multipolar InteractionsDocument11 pagesNon-Collinear Magnetism Due To Orbital Degeneracy and Multipolar InteractionsYoshio KuramotoNo ratings yet
- Allylic 1,3-Strain As A Controlling Factor in Stereoselective TransformationsDocument20 pagesAllylic 1,3-Strain As A Controlling Factor in Stereoselective TransformationsAlexander WetzelNo ratings yet
- Kaiser 1997 ApJ 477 982Document8 pagesKaiser 1997 ApJ 477 982giampieroNo ratings yet
- Theory of Antiferroelectric CrystalsDocument4 pagesTheory of Antiferroelectric CrystalsJJ SerraltaNo ratings yet
- Physical Organic Chemistry Chapter 4Document29 pagesPhysical Organic Chemistry Chapter 4MULUKEN TILAHUNNo ratings yet
- Artigo Do Corey Sobre Adição de LiAlH4 em Alcoóis PropargílicosDocument3 pagesArtigo Do Corey Sobre Adição de LiAlH4 em Alcoóis PropargílicosNatan FilippiNo ratings yet
- Huygens Institute - Royal Netherlands Academy of Arts and Sciences (KNAW)Document21 pagesHuygens Institute - Royal Netherlands Academy of Arts and Sciences (KNAW)JOSE JESUS OJEDA GARCIANo ratings yet
- Huang 2014Document5 pagesHuang 2014kenfackNo ratings yet
- SegertPhysRevLett 61 1329Document5 pagesSegertPhysRevLett 61 1329SiestaNo ratings yet
- Chalker FinalDocument44 pagesChalker FinalMuhammad Shifan JayadiNo ratings yet
- Artículo 4Document14 pagesArtículo 4Exlonk Gil PeláezNo ratings yet
- Photochemical Upconversion in Solution The Role of Oxygen and Magnetic Field ResponseDocument7 pagesPhotochemical Upconversion in Solution The Role of Oxygen and Magnetic Field ResponseSuman DeviNo ratings yet
- (NDWP1D) Schlagheck Epjd03Document15 pages(NDWP1D) Schlagheck Epjd03John LeónNo ratings yet
- 304-Article Text-485-1-10-20220226Document5 pages304-Article Text-485-1-10-20220226MasoodNo ratings yet
- An Infinite Family of 3d Floquet Topological ParamagnetsDocument13 pagesAn Infinite Family of 3d Floquet Topological Paramagnetshuevonomar05No ratings yet
- Question of Conservation In: Parity Weak InteractionsDocument5 pagesQuestion of Conservation In: Parity Weak InteractionsAmberNo ratings yet
- Ch. 3 Crystal: Field TheoryDocument1 pageCh. 3 Crystal: Field TheoryKamleshkekane1No ratings yet
- Geometric Frustration Inherent To The Trillium Lattice, A Sublattice of The B20 StructureDocument14 pagesGeometric Frustration Inherent To The Trillium Lattice, A Sublattice of The B20 Structureprissyben17No ratings yet
- Role of External Flow and Frame Invariance in Stochastic ThermodynamicsDocument4 pagesRole of External Flow and Frame Invariance in Stochastic ThermodynamicsahmadalsaiahNo ratings yet
- Rotational Tunneling in Solids: Theory Neutron Scattering.: W'estDocument12 pagesRotational Tunneling in Solids: Theory Neutron Scattering.: W'estsergiodominguesNo ratings yet
- Structure of CIF, and Exchange Studies On Some Halogen Fluorides Nuclear Magnetic Resonance1Document5 pagesStructure of CIF, and Exchange Studies On Some Halogen Fluorides Nuclear Magnetic Resonance1shajeshNo ratings yet
- Callen Onsager Transport PR1948Document10 pagesCallen Onsager Transport PR1948ibn_rafiNo ratings yet
- Mossbauer EffectDocument34 pagesMossbauer EffectRishi SankerNo ratings yet
- Flory 1958Document10 pagesFlory 1958Santiago ZapataNo ratings yet
- Nuclear Fusion - Energy For FutureDocument12 pagesNuclear Fusion - Energy For FuturedeepthiNo ratings yet
- The Conservation Orbital: of SymmetryDocument6 pagesThe Conservation Orbital: of SymmetrylovehopeNo ratings yet
- Oslonovich 2003Document12 pagesOslonovich 2003ferNo ratings yet
- P-Rez Et Al-2016-Chemistry - A European JournalDocument8 pagesP-Rez Et Al-2016-Chemistry - A European JournalLucilaNo ratings yet
- FullDocument5 pagesFullAbhishekChatterjeeNo ratings yet
- RevModPhys 36 31Document9 pagesRevModPhys 36 31Iuliuana MandruNo ratings yet
- Attractive Forces Between Long Saturated Chains at Short DistancesDocument15 pagesAttractive Forces Between Long Saturated Chains at Short DistancesAdriano HenriqueNo ratings yet
- Search Negative Parti Cles: Cosm RAYDocument5 pagesSearch Negative Parti Cles: Cosm RAYMónika BokorNo ratings yet
- The Journal of Chemical Physics Volume 20 Issue 4 1952 (Doi 10.1063 - 1.1700516) McClure, Donald S. - Spin-Orbit Interaction in Aromatic MoleculesDocument6 pagesThe Journal of Chemical Physics Volume 20 Issue 4 1952 (Doi 10.1063 - 1.1700516) McClure, Donald S. - Spin-Orbit Interaction in Aromatic MoleculesJustin MdNo ratings yet
- Weak Interactions and Nonconservation of Parity: Tsung Dao LeeDocument13 pagesWeak Interactions and Nonconservation of Parity: Tsung Dao LeesandugandhiNo ratings yet
- Dynamics of Hydrogen Bromide Dissolution in The Ground and Excited StatesDocument4 pagesDynamics of Hydrogen Bromide Dissolution in The Ground and Excited StatesTERESA CAROLINA CARCAMO CAMACHONo ratings yet
- Doping Dependence of Irreversibility Line in Y1 X 2010 Physica C SuperconduDocument7 pagesDoping Dependence of Irreversibility Line in Y1 X 2010 Physica C SuperconduBlue LlightningNo ratings yet
- Macromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976From EverandMacromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976B. SedláčekNo ratings yet
- Sample Paper-Ftre-2022-Class-X-P1-At+pcmDocument18 pagesSample Paper-Ftre-2022-Class-X-P1-At+pcmhipakin454100% (1)
- GRAFiTO REVIEWDocument15 pagesGRAFiTO REVIEWAgtc TandayNo ratings yet
- Metal ClusterDocument30 pagesMetal ClusterGokul KannanNo ratings yet
- Phetrattanarangsi 2017Document33 pagesPhetrattanarangsi 2017beki ayuuNo ratings yet
- 9 11Document1 page9 11hassan tariqNo ratings yet
- Chapter 1 - IntroductionDocument10 pagesChapter 1 - IntroductionroseNo ratings yet
- Dissertation Report PDFDocument70 pagesDissertation Report PDFRadhika KhandelwalNo ratings yet
- H S Removal TechnologyDocument31 pagesH S Removal Technologytaufik budiarjoNo ratings yet
- Syn Final Helen MDocument8 pagesSyn Final Helen Mapi-509765936No ratings yet
- Heterocyclic ChemistryDocument49 pagesHeterocyclic ChemistryVã RãNo ratings yet
- Pulp ProtectionDocument13 pagesPulp ProtectionmirfanulhaqNo ratings yet
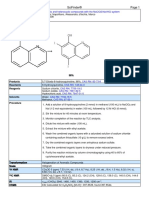
- Scifinder®: Mild and Efficient Iodination of Aromatic and Heterocyclic Compounds With The Naclo2/Nai/Hcl SystemDocument2 pagesScifinder®: Mild and Efficient Iodination of Aromatic and Heterocyclic Compounds With The Naclo2/Nai/Hcl SystemJesus Garcia PatiñoNo ratings yet
- 3e. Classification en 14700Document2 pages3e. Classification en 14700Nur Stya Auliya'uddinNo ratings yet
- Battery TechnologyDocument44 pagesBattery Technologyvishnu100% (1)
- Class 5 BleachingDocument20 pagesClass 5 BleachingAbdisadebele BedaneNo ratings yet
- Types of CompoundsDocument15 pagesTypes of CompoundsJonard PedrosaNo ratings yet
- 3,7-Dimethylpentadecane: A Novel Sex Pheromone Component From Leucoptera Sinuella (Lepidoptera: Lyonetiidae)Document10 pages3,7-Dimethylpentadecane: A Novel Sex Pheromone Component From Leucoptera Sinuella (Lepidoptera: Lyonetiidae)Heidy HerreraNo ratings yet
- A Novel Kind of Concrete Superplasticizer Based On LigniteDocument8 pagesA Novel Kind of Concrete Superplasticizer Based On Lignitemoustafa hadj-doulaNo ratings yet
- Parlon - PyroData 3Document3 pagesParlon - PyroData 3ricoNo ratings yet
- Introduction To General Organic and Biochemistry 12Th Edition Frederick A Bettelheim Full ChapterDocument67 pagesIntroduction To General Organic and Biochemistry 12Th Edition Frederick A Bettelheim Full Chapterronnie.ruch609100% (4)
- Why Choose Fiberglass: What Is Fiberglass Reinforced Polymer?Document2 pagesWhy Choose Fiberglass: What Is Fiberglass Reinforced Polymer?yanuar_adhiNo ratings yet
- Chapter 1 Cement Civil Engineering MaterialDocument12 pagesChapter 1 Cement Civil Engineering MaterialAnonymous 59kjvq4OLB100% (1)
- TUGAS G.5 GEOKIMIA (Simson Paulus Taifa)Document5 pagesTUGAS G.5 GEOKIMIA (Simson Paulus Taifa)Son TaifaNo ratings yet
- Journal of CO2 Utilization: 2 Ikhlas Ghiat, Tareq Al-AnsariDocument14 pagesJournal of CO2 Utilization: 2 Ikhlas Ghiat, Tareq Al-AnsariMuhammad Imran KhanNo ratings yet