You might also like
- SAP Group Reporting 1909Document28 pagesSAP Group Reporting 1909SUDIPTADATTARAY86% (7)
- 978-0393617542 Research MethodsDocument18 pages978-0393617542 Research MethodsReccebacaNo ratings yet
- The DAMA Guide To The Data Management Body of Knowledge - First EditionDocument430 pagesThe DAMA Guide To The Data Management Body of Knowledge - First Editionkakarotodesu100% (10)
- Inhibitors of MAO-A and MAO-B in Psychiatry and NeurologyDocument27 pagesInhibitors of MAO-A and MAO-B in Psychiatry and NeurologySergeyNo ratings yet
- Monoamine Oxidase Inhibitors in Parkinson's DiseaseDocument12 pagesMonoamine Oxidase Inhibitors in Parkinson's Diseaseelenac67100% (1)
- Deprenyl (Selegiline) : The History of Its Development and Pharmacological ActionDocument24 pagesDeprenyl (Selegiline) : The History of Its Development and Pharmacological ActionAs DvgNo ratings yet
- Biochemistry and Pharmacology of Reversible Inhibitors of MAO-Agents A Focus On MoclobemideDocument12 pagesBiochemistry and Pharmacology of Reversible Inhibitors of MAO-Agents A Focus On MoclobemideYoselin Milagros Chambi VelasquezNo ratings yet
- MoclobemideDocument26 pagesMoclobemideSkijanjeSrbijaNo ratings yet
- Current Place of Monoamine Oxidase Inhibitors in The Treatment of DepressionDocument9 pagesCurrent Place of Monoamine Oxidase Inhibitors in The Treatment of DepressiondanilomarandolaNo ratings yet
- Moclobemide: Evolution, Pharmacodynamic, and Pharmacokinetic PropertiesDocument26 pagesMoclobemide: Evolution, Pharmacodynamic, and Pharmacokinetic PropertiesGoretti ArvizuNo ratings yet
- Neuro Therapeutics 20102Document14 pagesNeuro Therapeutics 20102Louay KilaNo ratings yet
- 414 1685 1 PBDocument13 pages414 1685 1 PBMuhammad Sona KhanNo ratings yet
- Monoamine Transporters and Psychostimulant Addiction: Leonard L. Howell, Heather L. KimmelDocument22 pagesMonoamine Transporters and Psychostimulant Addiction: Leonard L. Howell, Heather L. KimmelDasein SerNo ratings yet
- Methylene Blue in The Treatment of Neuropsychiatric Disorders 2019Document7 pagesMethylene Blue in The Treatment of Neuropsychiatric Disorders 2019Agusti Caudet RoigNo ratings yet
- A Review: Hypothesis of Depression and Role of Antidepressant DrugsDocument4 pagesA Review: Hypothesis of Depression and Role of Antidepressant DrugsSuraj SawadekarNo ratings yet
- 4 6ZopicloneCritReviewDocument21 pages4 6ZopicloneCritReviewSven HasselNo ratings yet
- Pharmacological ReportsDocument10 pagesPharmacological ReportsFany Monterrubio LozanoNo ratings yet
- Mode of Action - ModafinilDocument16 pagesMode of Action - ModafinilellarifNo ratings yet
- Bioorganic & Medicinal Chemistry LettersDocument4 pagesBioorganic & Medicinal Chemistry LettersVinícius PiantaNo ratings yet
- PhenothiazineDocument11 pagesPhenothiazineB489Fadhlur RohmansyahNo ratings yet
- Development of Nociceptin ReceptorDocument44 pagesDevelopment of Nociceptin ReceptorAntônio Neto MachadoNo ratings yet
- Relationship Neurotransmitters Symptoms Major DepressiveDocument4 pagesRelationship Neurotransmitters Symptoms Major DepressiveLilian LópezNo ratings yet
- Nihms31302 PDFDocument15 pagesNihms31302 PDFTodor JelevNo ratings yet
- Is Immunomodulation by Opioid Drugs of Clinical RelevanceDocument5 pagesIs Immunomodulation by Opioid Drugs of Clinical RelevanceJack ShaaoNo ratings yet
- Adrenergic AgentsDocument7 pagesAdrenergic AgentsMuhamed ArsalanNo ratings yet
- Pharmacology of Medicinal Plants and Natural ProductsDocument116 pagesPharmacology of Medicinal Plants and Natural ProductsMichael MasengiNo ratings yet
- Theophylline - Recent Advances in The Understanding of Its Mode of Action and Uses in Clinical PracticeDocument9 pagesTheophylline - Recent Advances in The Understanding of Its Mode of Action and Uses in Clinical PracticeAndreiNo ratings yet
- MMJ Publication 2011Document4 pagesMMJ Publication 2011BijuKuriliKattleNo ratings yet
- A Systematic Review of Modafinil. Potential Clinical Uses & Mechanisms of ActionDocument13 pagesA Systematic Review of Modafinil. Potential Clinical Uses & Mechanisms of ActionMelissaNo ratings yet
- Nihms 485810Document17 pagesNihms 485810Joost WaasNo ratings yet
- 1 s2.0 S1359644613000949 MainDocument8 pages1 s2.0 S1359644613000949 MainPedro Henrique CesarNo ratings yet
- Laycock (2019) Opioid Mechanisms and Opioid DrugsDocument6 pagesLaycock (2019) Opioid Mechanisms and Opioid DrugsSofíaNo ratings yet
- Annals of The New York Academy of Sciences - July 1990 - GILMORE - The Role of Opioid Peptides in ImmunomodulationaDocument12 pagesAnnals of The New York Academy of Sciences - July 1990 - GILMORE - The Role of Opioid Peptides in ImmunomodulationaJack ShaaoNo ratings yet
- Eps Opioid WithdrawalDocument2 pagesEps Opioid WithdrawalPuneetNo ratings yet
- Molecules 26 03724Document21 pagesMolecules 26 03724Cioara IleanaNo ratings yet
- Anti Seizure Drugs 07Document27 pagesAnti Seizure Drugs 07ashokvishnoi29pNo ratings yet
- 05 Friedman 2020 Opiods ReviewDocument15 pages05 Friedman 2020 Opiods ReviewJorge GomezNo ratings yet
- Neer Man 2003Document4 pagesNeer Man 2003Tia RahayuNo ratings yet
- Running Head: Biochemistry Assignment 1Document6 pagesRunning Head: Biochemistry Assignment 1Fredrick LiyengaNo ratings yet
- Melatonin Hormone of The NightDocument3 pagesMelatonin Hormone of The NightFábio Yutani KosekiNo ratings yet
- MoleculesDocument10 pagesMoleculesWalid Ebid ElgammalNo ratings yet
- Anderzhanova 2013Document10 pagesAnderzhanova 2013ASHUTOSH KUMAR SinghNo ratings yet
- DKT 5988 1 Redacted Report of Michael Golding Ocr 10-31-2018 489 3Document1 pageDKT 5988 1 Redacted Report of Michael Golding Ocr 10-31-2018 489 3VokdadaNo ratings yet
- Opioid Chapter 2Document7 pagesOpioid Chapter 2Likoh TimothyNo ratings yet
- Molecular Mechanisms Underlying The Involvement of The S1R in Methamphetamine Mediated Microglia Polarization - Chao2017Document13 pagesMolecular Mechanisms Underlying The Involvement of The S1R in Methamphetamine Mediated Microglia Polarization - Chao2017nienminhNo ratings yet
- Background: 1.3-Benzothiazoles As Anti-Alzheimer's (AD) DiseaseDocument1 pageBackground: 1.3-Benzothiazoles As Anti-Alzheimer's (AD) DiseaseمنارNo ratings yet
- A Systematic Review of Modafinil: Potential Clinical Uses and Mechanisms of ActionDocument26 pagesA Systematic Review of Modafinil: Potential Clinical Uses and Mechanisms of ActionAnkit HasijaNo ratings yet
- The Biochemical Basis of Neuropharmacology 8th EditionDocument513 pagesThe Biochemical Basis of Neuropharmacology 8th EditionNeuro Iztacala100% (1)
- Neuroscience Letters: Laura Monni, Filippo Ghezzi, Silvia Corsini, Andrea NistriDocument6 pagesNeuroscience Letters: Laura Monni, Filippo Ghezzi, Silvia Corsini, Andrea NistriardhiNo ratings yet
- HypothesisDocument2 pagesHypothesisJomana MacalnasNo ratings yet
- NIHSS IndonesiaDocument17 pagesNIHSS IndonesiaBesta Arum BelaNo ratings yet
- DialoguesClinNeurosci 7 223Document8 pagesDialoguesClinNeurosci 7 223mantradaNo ratings yet
- Synthetic Isoflavones and Doping: A Novel Class of Aromatase Inhibitors?Document23 pagesSynthetic Isoflavones and Doping: A Novel Class of Aromatase Inhibitors?Nimra Naveed ShaikhNo ratings yet
- Badr Et Al, 2020Document10 pagesBadr Et Al, 2020UrwaTariqNo ratings yet
- Modafinil AnnotatedDocument13 pagesModafinil AnnotatedMistah RoflcopterNo ratings yet
- Journal of Neurochemistry - 2011 - Jafari - Structural Contributions of Antipsychotic Drugs To Their Therapeutic ProfilesDocument14 pagesJournal of Neurochemistry - 2011 - Jafari - Structural Contributions of Antipsychotic Drugs To Their Therapeutic Profilesjohnny chawNo ratings yet
- Monoamine Oxidase A and B Inhibitors in Parkins 2007 Handbook of Clinical Ne 2Document28 pagesMonoamine Oxidase A and B Inhibitors in Parkins 2007 Handbook of Clinical Ne 2eylul.irmak.tNo ratings yet
- PDF - Why Dont We Prescribe MAOIsDocument11 pagesPDF - Why Dont We Prescribe MAOIsSarwar BaigNo ratings yet
- Review (1) - 2004 - Tabakmna Et AlDocument11 pagesReview (1) - 2004 - Tabakmna Et Alshimonl3892No ratings yet
- Omcl0204 - 0181 - Synthesis of MescalineDocument10 pagesOmcl0204 - 0181 - Synthesis of MescalineOleksandr StorcheusNo ratings yet
- Side Effects of Anti Psychotic MedicationsDocument11 pagesSide Effects of Anti Psychotic MedicationsluciapopNo ratings yet
- Agomelatine MelatoninDocument18 pagesAgomelatine MelatoninSj SpNo ratings yet
- An Introduction to Mechanisms in Pharmacology and TherapeuticsFrom EverandAn Introduction to Mechanisms in Pharmacology and TherapeuticsNo ratings yet
- Machine Design Key 2014Document15 pagesMachine Design Key 2014SouvikDasNo ratings yet
- Mutant ChroniclesDocument3 pagesMutant ChroniclesZoth BernsteinNo ratings yet
- Trockel Flash Art 1987Document4 pagesTrockel Flash Art 1987cvg_geNo ratings yet
- Chemistry Bridging Course Lecture NotesDocument3 pagesChemistry Bridging Course Lecture NotesNNo ratings yet
- Expt Lipid - Influence of Bile in The Action of LipaseDocument3 pagesExpt Lipid - Influence of Bile in The Action of LipaseAngela CaguitlaNo ratings yet
- 18TE72 - Wireless Communication Experiential Learning ReportDocument10 pages18TE72 - Wireless Communication Experiential Learning ReportRitika BakshiNo ratings yet
- Tea Board of India PDFDocument18 pagesTea Board of India PDFDebasish RazNo ratings yet
- CM658 Time - MNGT - 10 - Getting Know The Class - NewNormDocument12 pagesCM658 Time - MNGT - 10 - Getting Know The Class - NewNormLee BañezNo ratings yet
- Water Cement RatioDocument5 pagesWater Cement RatioCastro FarfansNo ratings yet
- Za Deloitte Intelligent Mining InfographicDocument4 pagesZa Deloitte Intelligent Mining InfographicAgung SupriyantoNo ratings yet
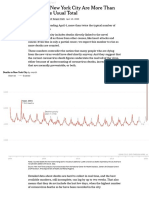
- Deaths in New York City Are More Than Double The Usual TotalDocument3 pagesDeaths in New York City Are More Than Double The Usual TotalRamón RuizNo ratings yet
- EKOFLUID PRODUCT SHEET FILOIL 6000 210x297 EN WEBDocument4 pagesEKOFLUID PRODUCT SHEET FILOIL 6000 210x297 EN WEBjunaidi.036No ratings yet
- Portal 2 PresDocument15 pagesPortal 2 Presapi-666608332No ratings yet
- GW1101-1DI (3IN1) DatasheetDocument6 pagesGW1101-1DI (3IN1) DatasheetGina HuachoNo ratings yet
- Analysis and Design of Suspended Buildings: Creative and Innovative Report-1Document14 pagesAnalysis and Design of Suspended Buildings: Creative and Innovative Report-1Nirmal RaviNo ratings yet
- Aurora National High School: Report On AttendanceDocument2 pagesAurora National High School: Report On AttendanceLimuel CaringalNo ratings yet
- UntitledDocument4 pagesUntitledapi-223522684No ratings yet
- Gopesh Obalappa Pilot ResumeDocument3 pagesGopesh Obalappa Pilot ResumeGopesh ObalappaNo ratings yet
- Armenotech PCIDSS AOCDocument13 pagesArmenotech PCIDSS AOCHakob ArakelyanNo ratings yet
- UNIABROAD PitchdeckDocument21 pagesUNIABROAD PitchdeckVikas MurulidharaNo ratings yet
- Predicates and ArgumentsDocument4 pagesPredicates and ArgumentsOanh NguyễnNo ratings yet
- Suricata User Guide: Release 4.1.0-DevDocument272 pagesSuricata User Guide: Release 4.1.0-DevDavid Simon Hoyos GonzalezNo ratings yet
- Guru Stotram-1Document5 pagesGuru Stotram-1Green WattNo ratings yet
- BarDocument1 pageBarJoannalyn Libo-onNo ratings yet
- REACH ArticlesDocument12 pagesREACH ArticlesChristian SugasttiNo ratings yet
- Sheet - PDF 3Document4 pagesSheet - PDF 3Nazar JabbarNo ratings yet
- EPB2.4 + V3f20 Installation - Start-Up ProcDocument30 pagesEPB2.4 + V3f20 Installation - Start-Up ProcBeltran Héctor75% (4)