You might also like
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Genetic Genie Methylation ProfileDocument6 pagesGenetic Genie Methylation ProfileadamNo ratings yet
- Summary Table Endocrine Review - OneDocument5 pagesSummary Table Endocrine Review - OneIndigoSilverNo ratings yet
- Las Heridas y Su Cicatrización: DermatologíaDocument6 pagesLas Heridas y Su Cicatrización: DermatologíaPaulyMartinezNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Perinatal and Developmental EpigeneticsFrom EverandPerinatal and Developmental EpigeneticsGarima SinghNo ratings yet
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Crecimiento y DesarrolloDocument41 pagesCrecimiento y DesarrolloJimmy15No ratings yet
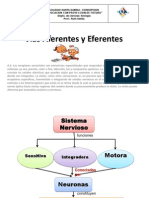
- s.n.4 (Vias Aferentes y Eferentes)Document24 pagess.n.4 (Vias Aferentes y Eferentes)tornes9262No ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5807)
- AACR 2022 Proceedings: Part A Online-Only and April 10From EverandAACR 2022 Proceedings: Part A Online-Only and April 10No ratings yet
- Starving Cancer Cells: Evidence-Based Strategies to Slow Cancer Progression: A Selection of Readings for Health Services ProvidersFrom EverandStarving Cancer Cells: Evidence-Based Strategies to Slow Cancer Progression: A Selection of Readings for Health Services ProvidersNo ratings yet
- Testes Psicológicos - Lista Por ÁreaDocument9 pagesTestes Psicológicos - Lista Por ÁreaLeonir Troscki67% (3)
- Map for ‘Drug and Food’ in Cancer Nutrition: Specific Nutrition Pathways During Chemotherapy for Patients with CancerFrom EverandMap for ‘Drug and Food’ in Cancer Nutrition: Specific Nutrition Pathways During Chemotherapy for Patients with CancerNo ratings yet
- Diagnosis and Management of Hereditary Cancer: Tabular-Based Clinical and Genetic AspectsFrom EverandDiagnosis and Management of Hereditary Cancer: Tabular-Based Clinical and Genetic AspectsNo ratings yet
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- Cellular Endocrinology in Health and DiseaseFrom EverandCellular Endocrinology in Health and DiseaseAlfredo Ulloa-AguirreNo ratings yet
- Clinical Genetics and Genomics of AgingFrom EverandClinical Genetics and Genomics of AgingJuan Carlos Gomez-VerjanNo ratings yet
- AACR 2016: Abstracts 1-2696From EverandAACR 2016: Abstracts 1-2696No ratings yet
- Next Generation Kinase Inhibitors: Moving Beyond the ATP Binding/Catalytic SitesFrom EverandNext Generation Kinase Inhibitors: Moving Beyond the ATP Binding/Catalytic SitesNo ratings yet
- Management of Urologic Cancer: Focal Therapy and Tissue PreservationFrom EverandManagement of Urologic Cancer: Focal Therapy and Tissue PreservationNo ratings yet
- AACR 2017 Proceedings: Abstracts 3063-5947From EverandAACR 2017 Proceedings: Abstracts 3063-5947No ratings yet
- AACR 2017 Proceedings: Abstracts 1-3062From EverandAACR 2017 Proceedings: Abstracts 1-3062No ratings yet
- Tumor Immune Microenvironment in Cancer Progression and Cancer TherapyFrom EverandTumor Immune Microenvironment in Cancer Progression and Cancer TherapyPawel KalinskiNo ratings yet
- Total Scar Management: From Lasers to Surgery for Scars, Keloids, and Scar ContracturesFrom EverandTotal Scar Management: From Lasers to Surgery for Scars, Keloids, and Scar ContracturesRei OgawaNo ratings yet
- Cancer Regional Therapy: HAI, HIPEC, HILP, ILI, PIPAC and BeyondFrom EverandCancer Regional Therapy: HAI, HIPEC, HILP, ILI, PIPAC and BeyondNo ratings yet
- Targeted Therapies for Lung CancerFrom EverandTargeted Therapies for Lung CancerRavi SalgiaNo ratings yet
- Tissue Engineering and Regeneration in Dentistry: Current StrategiesFrom EverandTissue Engineering and Regeneration in Dentistry: Current StrategiesRachel J. WaddingtonNo ratings yet
- Hormones and Embryonic Development: Advances in the BiosciencesFrom EverandHormones and Embryonic Development: Advances in the BiosciencesG. RaspéNo ratings yet
- PAKs, RAC/CDC42 (p21)-activated Kinases: Towards the Cure of Cancer and Other PAK-dependent DiseasesFrom EverandPAKs, RAC/CDC42 (p21)-activated Kinases: Towards the Cure of Cancer and Other PAK-dependent DiseasesHiroshi MarutaNo ratings yet
- Stem Cells and Cell Signalling in Skeletal MyogenesisFrom EverandStem Cells and Cell Signalling in Skeletal MyogenesisNo ratings yet
- Molecular and Cellular Biology of Pathogenic TrypanosomatidsFrom EverandMolecular and Cellular Biology of Pathogenic TrypanosomatidsNo ratings yet
- Novel Sensitizing Agents for Therapeutic Anti-EGFR AntibodiesFrom EverandNovel Sensitizing Agents for Therapeutic Anti-EGFR AntibodiesShi HuNo ratings yet
- Extracellular Targeting of Cell Signaling in Cancer: Strategies Directed at MET and RON Receptor Tyrosine Kinase PathwaysFrom EverandExtracellular Targeting of Cell Signaling in Cancer: Strategies Directed at MET and RON Receptor Tyrosine Kinase PathwaysJames W. JanetkaNo ratings yet
- Molecular Basis of Biological Degradative processesFrom EverandMolecular Basis of Biological Degradative processesRichard BerlinNo ratings yet
- Xenobiotic Regulation of Estrogen and Progesterone Receptor - Mediated Gene ExpressionFrom EverandXenobiotic Regulation of Estrogen and Progesterone Receptor - Mediated Gene ExpressionNo ratings yet
- A Patient's Guide to Cancer: Understanding the Causes and Treatments of a Complex DiseaseFrom EverandA Patient's Guide to Cancer: Understanding the Causes and Treatments of a Complex DiseaseNo ratings yet
- Jose Enrique Romero RamosDocument58 pagesJose Enrique Romero RamosAlvaro GiraldoNo ratings yet
- Trastornos Del Estado de AnimoDocument31 pagesTrastornos Del Estado de AnimoDeisy Mamani TNo ratings yet
- GPC AsmaDocument20 pagesGPC AsmaRolón SpuakNo ratings yet
- Biology of T Cruzi Annurev - Mi.27.100173.002023 73Document36 pagesBiology of T Cruzi Annurev - Mi.27.100173.002023 73mflorezmNo ratings yet
- 1 Envejecimiento de Los Organos de Los SentidosDocument51 pages1 Envejecimiento de Los Organos de Los SentidosRoberto SolaresNo ratings yet
- Funciones Vitales Escala Animal CirculacionDocument7 pagesFunciones Vitales Escala Animal Circulacionvoldur12No ratings yet
- Embriología Sistema Urinario PDFDocument17 pagesEmbriología Sistema Urinario PDFBryan Lema MasacheNo ratings yet
- UNIDAD 3-Ganglio LinfáticoDocument48 pagesUNIDAD 3-Ganglio LinfáticoJorge LucasNo ratings yet
- E Spermogram ADocument17 pagesE Spermogram AJulianoPereiraNo ratings yet
- MetamizolDocument20 pagesMetamizoljose de jesus mares sanchezNo ratings yet
- 1 Ano Quimica Da VidaDocument24 pages1 Ano Quimica Da VidaDaniel ViníciusNo ratings yet
- Case Study On GMOsDocument11 pagesCase Study On GMOsroselle azucena100% (1)
- Portada Tesis Esclerosis CorrecionDocument12 pagesPortada Tesis Esclerosis CorrecionLisbeth ParraNo ratings yet
- Lista 1 ALTERNATIVA AjeitandoDocument7 pagesLista 1 ALTERNATIVA AjeitandoKeiciane Canabarro Drehmer MarquesNo ratings yet
- Mucuna PresentaciónDocument14 pagesMucuna PresentaciónEdgar Ricardo Cruz HernándezNo ratings yet
- Lab Gen Parcial 2Document43 pagesLab Gen Parcial 2Nadia Karime Silva FloresNo ratings yet
- Aditivos AlimentariosDocument12 pagesAditivos AlimentariosXavier Ramos FloresNo ratings yet
- DiencefaloDocument6 pagesDiencefaloAnais Velasquez MuñozNo ratings yet
- Retard Mitotique Et Maintien de L'intégrité ChromosomiqueDocument28 pagesRetard Mitotique Et Maintien de L'intégrité ChromosomiqueAnis Wins50% (2)
- Human Genetics Unit Test Study Guide - 09-10Document5 pagesHuman Genetics Unit Test Study Guide - 09-10Tina HowitsonNo ratings yet
- Cruces Con Drosophila Melanogaster CruceDocument9 pagesCruces Con Drosophila Melanogaster CruceYina Saldaña GaribayNo ratings yet
- Tema 6 - Afectividad Negativa y PersonalidadDocument10 pagesTema 6 - Afectividad Negativa y PersonalidadJuanokNo ratings yet
- Alternaria SolaniDocument7 pagesAlternaria SolanimilkaNo ratings yet
- Pathogenesis of Group A Streptococcal InfectionsDocument59 pagesPathogenesis of Group A Streptococcal InfectionsHerdwin Limas IINo ratings yet