You might also like
- CAE Practice Test Plus 2Document242 pagesCAE Practice Test Plus 2Enrique MC100% (2)
- Diabetes InsipidusDocument21 pagesDiabetes InsipidusMoni RethNo ratings yet
- Pharmacokinetics of AnestheticsDocument5 pagesPharmacokinetics of AnestheticsFairysparklesNo ratings yet
- CKD Product - BD PackageDocument33 pagesCKD Product - BD PackageAhmedNo ratings yet
- Diabetes InsipidaDocument15 pagesDiabetes Insipidapaulito_delgadoNo ratings yet
- NCBI Bookshelf-Diabetes InsipidusDocument44 pagesNCBI Bookshelf-Diabetes InsipidusDiah Pradnya ParamitaNo ratings yet
- Best Practice & Research Clinical Endocrinology & MetabolismDocument11 pagesBest Practice & Research Clinical Endocrinology & Metabolismpaulito_delgadoNo ratings yet
- Case Report - Diabetes InsipidusDocument4 pagesCase Report - Diabetes InsipidusSACHI DANIELLA TATOYNo ratings yet
- Pediatric Diabetes InsipidusDocument11 pagesPediatric Diabetes InsipidusVictor Matias BarriosNo ratings yet
- Diabete InsipidaDocument20 pagesDiabete InsipidaLuciana RafaelNo ratings yet
- Diabetes InsipidusDocument12 pagesDiabetes InsipidusAbhijeetNo ratings yet
- Treatment of Neurohypophyseal Diabetes Insipidus - Oiso YDocument10 pagesTreatment of Neurohypophyseal Diabetes Insipidus - Oiso Yshintia novotnaNo ratings yet
- Diabetes Insipida. Tratamiento NeurohipofisiarioDocument10 pagesDiabetes Insipida. Tratamiento NeurohipofisiarioJesus OsorioNo ratings yet
- GBSDocument21 pagesGBSCherr NollNo ratings yet
- D InsipidaDocument3 pagesD InsipidaAndrea Valenzuela AedoNo ratings yet
- Lab 5 Diabetes InsipidusDocument6 pagesLab 5 Diabetes InsipidusLisa EkapratiwiNo ratings yet
- Diagnosis of Polyuria and Diabetes Insipidus - UP To DATEDocument15 pagesDiagnosis of Polyuria and Diabetes Insipidus - UP To DATELuis Fernando Arias VillalobosNo ratings yet
- Diabetes InsipidusDocument17 pagesDiabetes InsipidusSan Siddz100% (2)
- Trastornos NeurohipofisisDocument11 pagesTrastornos NeurohipofisisIx Chel GlezNo ratings yet
- Nephrotic Syndrome in Children-LectureDocument52 pagesNephrotic Syndrome in Children-LectureLubinda SitaliNo ratings yet
- Diabetes InsipidusDocument18 pagesDiabetes InsipidusElena Patricia GonzálezNo ratings yet
- Nephrogenic Diabetes InsipidusDocument10 pagesNephrogenic Diabetes InsipidusJapheth SalvanNo ratings yet
- Syndromeofinappropriate Antidiuresis: Michael L. MoritzDocument18 pagesSyndromeofinappropriate Antidiuresis: Michael L. MoritznelidayslasNo ratings yet
- Diabetes Insipidus: Presented By-Akansha Student, B.sc. Nursing 3rd Semester Con, DR - RmlimsDocument12 pagesDiabetes Insipidus: Presented By-Akansha Student, B.sc. Nursing 3rd Semester Con, DR - Rmlimsakanshaa32000No ratings yet
- A Copeptin-Based Classification of The Osmoregulatory DefectDocument15 pagesA Copeptin-Based Classification of The Osmoregulatory DefectDEWINo ratings yet
- Diabetes InsipidusDocument10 pagesDiabetes InsipidussoniaNo ratings yet
- Systemic Effects of Perinatal AsphyxiaDocument10 pagesSystemic Effects of Perinatal AsphyxiaJaner Banos100% (1)
- Metabolik EncelophatyDocument8 pagesMetabolik EncelophatyfkiaNo ratings yet
- Pa Tho Physiology of Diabetes InsipidusDocument4 pagesPa Tho Physiology of Diabetes InsipidusteynsNo ratings yet
- Careplan 2Document11 pagesCareplan 2JulieRn2beNo ratings yet
- Diabetes Insipidus HandoutDocument4 pagesDiabetes Insipidus HandoutllianNo ratings yet
- GRP4 - Ppt-Diabetes InsipidusDocument12 pagesGRP4 - Ppt-Diabetes InsipidusNicole Villanueva, BSN - Level 3A100% (2)
- Means Brain Disease, Damage, or Malfunction. The Causes Metabolic Problems, Toxins, Drugs, Physiologic Changes, Trauma, and Other CausesDocument9 pagesMeans Brain Disease, Damage, or Malfunction. The Causes Metabolic Problems, Toxins, Drugs, Physiologic Changes, Trauma, and Other CausesMacey MalonzoNo ratings yet
- Diabetes InsipidusDocument7 pagesDiabetes Insipiduspaul_stefenson10No ratings yet
- Drug StudiesDocument16 pagesDrug Studiesvitcloud23100% (2)
- Diabetes Insipidus: Presented By-Akansha Student, B.sc. Nursing 3rd Semester Con, DR - RmlimsDocument12 pagesDiabetes Insipidus: Presented By-Akansha Student, B.sc. Nursing 3rd Semester Con, DR - RmlimsAkansha gandharvNo ratings yet
- Sands PDFDocument10 pagesSands PDFAsmaulKhusnaNo ratings yet
- The Management of Vasovagal Syncope: R.A. Kenny and T. McnicholasDocument8 pagesThe Management of Vasovagal Syncope: R.A. Kenny and T. McnicholasKat ZNo ratings yet
- Juraj Artner - Cancer and Neoplasia FactsheetDocument11 pagesJuraj Artner - Cancer and Neoplasia FactsheetBogdan SoltuzuNo ratings yet
- Clinical Manifestations and Causes of Central Diabetes InsipidusDocument12 pagesClinical Manifestations and Causes of Central Diabetes InsipidusTessa AcostaNo ratings yet
- UNIT-V PharmacologyofDrugsactingonRenalsystem AntidiuerticsDocument10 pagesUNIT-V PharmacologyofDrugsactingonRenalsystem AntidiuerticsMisganaw Abate AbateNo ratings yet
- Clinical Disorders Excess Vasopressin/syndrome of Inappropriate Antidiuretic HormoneDocument5 pagesClinical Disorders Excess Vasopressin/syndrome of Inappropriate Antidiuretic HormoneDirgantari PademmeNo ratings yet
- Birth Asphyxia: DefinitionDocument5 pagesBirth Asphyxia: DefinitionDella FerginaNo ratings yet
- Non-Hodgkin's LymphomaDocument17 pagesNon-Hodgkin's LymphomaMiche DairoNo ratings yet
- Rehab Elective PresentationDocument21 pagesRehab Elective PresentationTahmina ShepuNo ratings yet
- Diabetes InsipidusDocument5 pagesDiabetes InsipidusFazalJaturieNo ratings yet
- Diabetes InsipidusDocument3 pagesDiabetes InsipidusCASE STUDYNo ratings yet
- Uptdate PediatricsDocument451 pagesUptdate PediatricsdrsaleemNo ratings yet
- A Review On Diabetic Neuropathy Complications and TreatmentDocument8 pagesA Review On Diabetic Neuropathy Complications and TreatmentPrapawee PhromsungwongNo ratings yet
- HydropsDocument13 pagesHydropsalejandro1122No ratings yet
- Causes and Treatment of Nausea and Vomiting: Aaron Bhakta and Rishi GoelDocument7 pagesCauses and Treatment of Nausea and Vomiting: Aaron Bhakta and Rishi GoelVini SasmitaNo ratings yet
- Deficiencies of Vasopressin Secretion and ActionDocument71 pagesDeficiencies of Vasopressin Secretion and Actionbiniam MesfinNo ratings yet
- JPM 13 00140 v2Document17 pagesJPM 13 00140 v2YeseniaNo ratings yet
- 28sici 291096 9136 28199802 2915 3A2 3C97 3A 3aaid Dia523 3e3.0.co 3B2 5 PDFDocument16 pages28sici 291096 9136 28199802 2915 3A2 3C97 3A 3aaid Dia523 3e3.0.co 3B2 5 PDFHesbon MomanyiNo ratings yet
- Hypernatremia NEJM 2000Document8 pagesHypernatremia NEJM 2000BenjamÍn Alejandro Ruiz ManzanoNo ratings yet
- 2010 New Aspects in The Pathogenesis, Prevention and Treatment Hyponatremic Encephalopathy ChildrenDocument14 pages2010 New Aspects in The Pathogenesis, Prevention and Treatment Hyponatremic Encephalopathy ChildrenJosibel OcantoNo ratings yet
- CVA NotesDocument21 pagesCVA NotesVaibhav BhatiaNo ratings yet
- Drug StudyDocument8 pagesDrug StudyLj PingoyNo ratings yet
- Posterior Reversible Encephalopathy SyndromeDocument7 pagesPosterior Reversible Encephalopathy Syndromemohana reddyNo ratings yet
- Care Plan NRS 108Document10 pagesCare Plan NRS 108Don Prem LegaspiNo ratings yet
- Diabetes INSIPIDUSDocument6 pagesDiabetes INSIPIDUSavinash dhameriya100% (1)
- Nephrotic Syndrome FinalDocument5 pagesNephrotic Syndrome FinalAaron AntonioNo ratings yet
- Epstein Barr VirusDocument13 pagesEpstein Barr VirusDianaLorenaNo ratings yet
- Cavitatea ToracicaDocument35 pagesCavitatea ToracicaDianaLorenaNo ratings yet
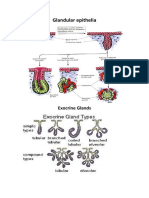
- Glandular Epithelia: Exocrine GlandsDocument6 pagesGlandular Epithelia: Exocrine GlandsDianaLorenaNo ratings yet
- TierneyDocument9 pagesTierneyAna MoraisNo ratings yet
- Msa Eu Us CTD List Oct 2022Document26 pagesMsa Eu Us CTD List Oct 2022Ning KevinNo ratings yet
- Practice Problems Answer KeyDocument4 pagesPractice Problems Answer KeyihuyphanNo ratings yet
- Management of Hypertension: Affan Syafiqi - Nurul Husna - Audi RahmanDocument77 pagesManagement of Hypertension: Affan Syafiqi - Nurul Husna - Audi RahmanRavi K. ShuklaNo ratings yet
- Pharmacy LabelDocument1 pagePharmacy LabelJannel MagalonaNo ratings yet
- AntibioticsDocument16 pagesAntibioticsVincent Raphael Baraan AcostaNo ratings yet
- Introduction To Pharmacognosy: Prepared byDocument47 pagesIntroduction To Pharmacognosy: Prepared byZahwa Syuhada FitriNo ratings yet
- GLAUCOMADocument10 pagesGLAUCOMAcarls burg a. resurreccionNo ratings yet
- Drug Infusion Guide For SORT PDFDocument5 pagesDrug Infusion Guide For SORT PDFAli HasanNo ratings yet
- Serous Otitis Media Clinical and Therapeutic ConsiDocument5 pagesSerous Otitis Media Clinical and Therapeutic Consiheidi leeNo ratings yet
- Engineering Ethics Case StudyDocument5 pagesEngineering Ethics Case Studyijaz aliNo ratings yet
- Formulation and Evaluation of Vilazodone Sublingual Tablets by Using Lyophilization TechniqueDocument9 pagesFormulation and Evaluation of Vilazodone Sublingual Tablets by Using Lyophilization Techniquealamia pharmNo ratings yet
- Hallucination - WikipediaDocument91 pagesHallucination - Wikipediakarthiksaran594No ratings yet
- Graylands Hospital Drug Bulletin: Using Lithium SafelyDocument4 pagesGraylands Hospital Drug Bulletin: Using Lithium Safelyanjali aroraNo ratings yet
- Internal Medicine: Dr. Nindyasari Diajeng Larasati Tim UKMPPD FKU MalahayatiDocument209 pagesInternal Medicine: Dr. Nindyasari Diajeng Larasati Tim UKMPPD FKU MalahayatiSilvi Qiro'atul AiniNo ratings yet
- Marketing Ayurveda-Recent Trends: Currently-CONSULTANT HR & Administration, FDCM ESSELWORLD GOREWADA ZOO Pvt. LTDDocument27 pagesMarketing Ayurveda-Recent Trends: Currently-CONSULTANT HR & Administration, FDCM ESSELWORLD GOREWADA ZOO Pvt. LTDUday DokrasNo ratings yet
- The Role of Medications in Weight LossDocument14 pagesThe Role of Medications in Weight LossWareesa JamshedNo ratings yet
- Unit III Drug EducationDocument12 pagesUnit III Drug EducationAbigail ParungaoNo ratings yet
- Chapter 34 - Anti-Inflammatory, Antipyretic, and Analgesic Agents Pharmacotherapy of GoutDocument57 pagesChapter 34 - Anti-Inflammatory, Antipyretic, and Analgesic Agents Pharmacotherapy of Goutsunita thakurNo ratings yet
- Premedication of The Child Undergoing Surgery: C. A R. HDocument12 pagesPremedication of The Child Undergoing Surgery: C. A R. HFatty MaulidiraNo ratings yet
- Drug Study For Losartan PotassiumDocument2 pagesDrug Study For Losartan PotassiumChryst Louise SaavedraNo ratings yet
- Natural Medicines For Psychotic DisordersDocument21 pagesNatural Medicines For Psychotic DisordersNath miaounNo ratings yet
- Ethnomedicinal Uses, Biological Activities, and Toxicity of Voacanga Africana Stapf Ex Scott-ElliotDocument19 pagesEthnomedicinal Uses, Biological Activities, and Toxicity of Voacanga Africana Stapf Ex Scott-ElliotMauricio CMNo ratings yet
- Buzzy With PhlebotomyDocument8 pagesBuzzy With PhlebotomyYusi nursiamNo ratings yet
- ACLS Drug Therapy RevisedDocument4 pagesACLS Drug Therapy RevisedpaveethrahNo ratings yet
- Formulir Algoritma NaranjoDocument1 pageFormulir Algoritma NaranjoSal MaiNo ratings yet
- Geriatric Giants Iwal 1 2020Document58 pagesGeriatric Giants Iwal 1 2020selymariaNo ratings yet
- Antibacterial Activity of Formulated Averrhoa Bilimbi (Kamias) Hand Sanitizer Gel On Staphylococcus AureusDocument13 pagesAntibacterial Activity of Formulated Averrhoa Bilimbi (Kamias) Hand Sanitizer Gel On Staphylococcus AureusMarvel Felicity Rosell ArmestoNo ratings yet