You might also like
- Nanocrystalline Materials: Their Synthesis-Structure-Property Relationships and ApplicationsFrom EverandNanocrystalline Materials: Their Synthesis-Structure-Property Relationships and ApplicationsNo ratings yet
- DFT Studies of Lead-Free Perovskite Material for Solar CellsDocument21 pagesDFT Studies of Lead-Free Perovskite Material for Solar CellsMine ConfessionNo ratings yet
- 10.1038@s41578 019 0080 9Document20 pages10.1038@s41578 019 0080 9Karl ZeiessNo ratings yet
- Development of Stable and Optimized Bandgap Halide Chalcogenide Perovskite Materials For Photovoltaic ApplicationsDocument8 pagesDevelopment of Stable and Optimized Bandgap Halide Chalcogenide Perovskite Materials For Photovoltaic ApplicationsKIU PUBLICATION AND EXTENSIONNo ratings yet
- Ahmad 2021Document11 pagesAhmad 2021Rana MaazNo ratings yet
- 6-CsSnX3 (X I, BR, CL)Document7 pages6-CsSnX3 (X I, BR, CL)Muhammad UsmanNo ratings yet
- Advs 201700471Document15 pagesAdvs 201700471Alicja AnuszkiewiczNo ratings yet
- Oxygen Transport in Higher-Order Ruddlesden-Popper Phase MaterialsDocument10 pagesOxygen Transport in Higher-Order Ruddlesden-Popper Phase MaterialsrajanadarajanNo ratings yet
- RSC AdvancesDocument18 pagesRSC Advancesvijayamathubalan pandyNo ratings yet
- Nano Energy: ReviewDocument14 pagesNano Energy: ReviewSheharyar MunirNo ratings yet
- Zhang2017 Article Low-DimensionalHalidePerovskit PDFDocument26 pagesZhang2017 Article Low-DimensionalHalidePerovskit PDFHanry SkywalkerNo ratings yet
- Nitrogen Embedded Small Molecule Semiconducting Materials Effec 2019 Dyes ADocument8 pagesNitrogen Embedded Small Molecule Semiconducting Materials Effec 2019 Dyes Aمحمد ريزقيNo ratings yet
- Thesis On Spinel FerritesDocument9 pagesThesis On Spinel Ferritesisabelleonorpaterson100% (2)
- Journal of Alloys and Compounds: Microwave Dielectric Properties of Lanthanum Based Complex PerovskitesDocument12 pagesJournal of Alloys and Compounds: Microwave Dielectric Properties of Lanthanum Based Complex PerovskitesDiki AriNo ratings yet
- 1-s2.0-S0254058424000713-mainDocument33 pages1-s2.0-S0254058424000713-mainCaptain AmericaNo ratings yet
- Porous La Doped NiO Microsphers For SCDocument8 pagesPorous La Doped NiO Microsphers For SCM M EhsanNo ratings yet
- 1 s2.0 S2589965119300698 MainDocument20 pages1 s2.0 S2589965119300698 MainLogeswary FiterNo ratings yet
- Optical and Stability Studies of Lead Bromide Perovskite NanocrystalsDocument17 pagesOptical and Stability Studies of Lead Bromide Perovskite NanocrystalsSree HarishNo ratings yet
- Perovskite Solar CellsDocument33 pagesPerovskite Solar CellsArya KeserciNo ratings yet
- 2D Ruddlesden-Popper Perovskites For OptoelectronicsDocument15 pages2D Ruddlesden-Popper Perovskites For Optoelectronicskakagermino23No ratings yet
- 1 s2.0 S0925838816315341 MainDocument7 pages1 s2.0 S0925838816315341 MainŸāssēr BoudræãNo ratings yet
- High-Throughput Computational Design of HalideDocument22 pagesHigh-Throughput Computational Design of Halidejose manuel acosta saavedraNo ratings yet
- Ceramics International: A B A A ADocument10 pagesCeramics International: A B A A AM Fahad BhopalNo ratings yet
- A New Class of Perovskite HEOsDocument5 pagesA New Class of Perovskite HEOsrambabu surampallyNo ratings yet
- Nanoscale High-K Dielectrics For Junctionless Nanowire Transistor For Drain Current AnalysisDocument8 pagesNanoscale High-K Dielectrics For Junctionless Nanowire Transistor For Drain Current AnalysisHarsh SinghNo ratings yet
- Yuetal 2017 J. Phys. D Appl. Phys. 10.1088 1361-6463 Aa8beaDocument29 pagesYuetal 2017 J. Phys. D Appl. Phys. 10.1088 1361-6463 Aa8beaSatyam ShindeNo ratings yet
- Materials Today: Proceedings: L. Renuka, Preeti Mishra, Y.S. Vidya, G. Banuprakash, K.S. Anantharaju, K.N. HarishDocument6 pagesMaterials Today: Proceedings: L. Renuka, Preeti Mishra, Y.S. Vidya, G. Banuprakash, K.S. Anantharaju, K.N. HarishJuancho PachonNo ratings yet
- 1 s2.0 S0925838822039482 MainDocument9 pages1 s2.0 S0925838822039482 MainBest WishesNo ratings yet
- Chakraborty BerryDocument8 pagesChakraborty Berrytapnath_45272029No ratings yet
- 2009 Nat. Mater. Metal Halide Perovskites For Light-Emitting DiodesDocument12 pages2009 Nat. Mater. Metal Halide Perovskites For Light-Emitting Diodes이규형No ratings yet
- 1 s2.0 S0038092X22003528 MainDocument11 pages1 s2.0 S0038092X22003528 MainAamir ShafiqueNo ratings yet
- 2 NanoscaleDocument11 pages2 Nanoscalea s m mosabbirNo ratings yet
- T Idhar 2014Document8 pagesT Idhar 2014kmensahdaNo ratings yet
- High Entropy Spinel-Structure Oxide For Electrochemical ApplicationDocument8 pagesHigh Entropy Spinel-Structure Oxide For Electrochemical ApplicationAlejadra ManotasNo ratings yet
- 1-NaSrF3 and NaZnF3Document7 pages1-NaSrF3 and NaZnF3Muhammad UsmanNo ratings yet
- Challenges and Strategies Toward Long-Term Stability of Lead-Free Tin-Based Perovskite Solar CellsDocument14 pagesChallenges and Strategies Toward Long-Term Stability of Lead-Free Tin-Based Perovskite Solar Cellsamal shokryNo ratings yet
- Reduced Size Carbon Energy - 2023 - Lan - Preparation and Promising Optoelectronic Applications of Lead Halide Perovskite PatternedDocument25 pagesReduced Size Carbon Energy - 2023 - Lan - Preparation and Promising Optoelectronic Applications of Lead Halide Perovskite PatternedP WangNo ratings yet
- Ruqiya HeliyonDocument21 pagesRuqiya HeliyonMazhar Amjad GilaniNo ratings yet
- ArtDocument6 pagesArtogtorresdNo ratings yet
- Electrochimica Acta - 2016 PDFDocument9 pagesElectrochimica Acta - 2016 PDFRimiNo ratings yet
- MP Review PaperDocument10 pagesMP Review PaperMUHAMMAD USMAN Mechanical Pesh.-Batch 20No ratings yet
- Energy: Vishal Shrivastav, Shashank Sundriyal, Umesh K. Tiwari, Ki-Hyun Kim, Akash DeepDocument12 pagesEnergy: Vishal Shrivastav, Shashank Sundriyal, Umesh K. Tiwari, Ki-Hyun Kim, Akash DeepKishoreBabu SNo ratings yet
- Materials 16 02290Document15 pagesMaterials 16 02290Abdellatif EL HABIBNo ratings yet
- Heylion JanushexafluorocyclohexaneDocument14 pagesHeylion JanushexafluorocyclohexaneMazhar Amjad GilaniNo ratings yet
- N03-SID5-Journal of The American Ceramic Society - 2012 - Zhang - Structural Evolving Sequence and Porous Ba6Zr2Nb8O30 FerroelectricDocument6 pagesN03-SID5-Journal of The American Ceramic Society - 2012 - Zhang - Structural Evolving Sequence and Porous Ba6Zr2Nb8O30 Ferroelectricton nu thanh phuongNo ratings yet
- In Depth Study On Growth Aspects and Characteristic Properties of Semiorganic Nonlinear Optical Crystal 4-Dimethylaminopyridine Copper ChlorideDocument8 pagesIn Depth Study On Growth Aspects and Characteristic Properties of Semiorganic Nonlinear Optical Crystal 4-Dimethylaminopyridine Copper ChlorideOnime No IchinoseNo ratings yet
- JETNSRCarrero 2017Document16 pagesJETNSRCarrero 2017Pratikshya PriyadarshiniNo ratings yet
- A Facile and Effective Strategy To Synthesize OrthDocument7 pagesA Facile and Effective Strategy To Synthesize OrthNguyễn Phương Thảo .k47b.hoáNo ratings yet
- O-Coordinated W-Mo Dual-Atom CatalystDocument14 pagesO-Coordinated W-Mo Dual-Atom CatalystdebmallyNo ratings yet
- Emerging Solar Cell PDFDocument23 pagesEmerging Solar Cell PDFJaffar LoneNo ratings yet
- Journal of Physics and Chemistry of Solids: 3 2 9 Zia Ur Rehman, M. Awais Rehman, Hamna Chaudhry, Muhammad AwaisDocument8 pagesJournal of Physics and Chemistry of Solids: 3 2 9 Zia Ur Rehman, M. Awais Rehman, Hamna Chaudhry, Muhammad AwaisMuhammad AwaisNo ratings yet
- Electrochimica Acta: Richa Pandey, Nan Jian, Alexandra Inberg, Richard E. Palmer, Yosi Shacham-DiamandDocument7 pagesElectrochimica Acta: Richa Pandey, Nan Jian, Alexandra Inberg, Richard E. Palmer, Yosi Shacham-Diamandbib123456789huNo ratings yet
- Zinc Ferrite Anchored Multiwalled Carbon Nanotubes For High-Performance Supercapacitor ApplicationsDocument6 pagesZinc Ferrite Anchored Multiwalled Carbon Nanotubes For High-Performance Supercapacitor Applicationsvijayamathubalan pandyNo ratings yet
- Ma2019 DIPAB Based 2D Large in Plane PsDocument6 pagesMa2019 DIPAB Based 2D Large in Plane Pssai prashanthNo ratings yet
- Ece Perovskite Solar CellsDocument18 pagesEce Perovskite Solar Cellsnirmala M.S.No ratings yet
- Srep 28847Document7 pagesSrep 28847Roha RohaNo ratings yet
- Ece Perovskite Solar CellsDocument18 pagesEce Perovskite Solar CellsNew AccNo ratings yet
- 1 s2.0 S0925346722006462 MainDocument8 pages1 s2.0 S0925346722006462 MainlahcenoviNo ratings yet
- Diamond and Related MaterialsDocument9 pagesDiamond and Related MaterialsShahid RamayNo ratings yet
- Heliyon - Giant NLO Response and Deep Ultraviolet Transparency of DualDocument12 pagesHeliyon - Giant NLO Response and Deep Ultraviolet Transparency of DualMazhar Amjad GilaniNo ratings yet
- Article 1 NouveauDocument10 pagesArticle 1 NouveauMaroc EcoloadNo ratings yet
- Correabaena 2017Document18 pagesCorreabaena 2017Maroc EcoloadNo ratings yet
- Business Kpi Dashboard Showing Average Revenue and CLVDocument6 pagesBusiness Kpi Dashboard Showing Average Revenue and CLVMaroc EcoloadNo ratings yet
- 10 1016@j Solener 2021 03 005Document23 pages10 1016@j Solener 2021 03 005Maroc EcoloadNo ratings yet
- Investigation of Thermal Properties of Alumina Based NanofluidsDocument3 pagesInvestigation of Thermal Properties of Alumina Based NanofluidsMaroc EcoloadNo ratings yet
- Article 15Document18 pagesArticle 15Maroc EcoloadNo ratings yet
- Catalysts 12 00964Document11 pagesCatalysts 12 00964Maroc EcoloadNo ratings yet
- Exercise 3c ChemistryDocument6 pagesExercise 3c Chemistryapi-533545229No ratings yet
- Group - 2 Practice Review QuestionsDocument5 pagesGroup - 2 Practice Review Questionsdosibo2378No ratings yet
- Kronnig Penney ModelDocument16 pagesKronnig Penney ModelAna PrpicNo ratings yet
- 1B Worksheet 10 EMFDocument9 pages1B Worksheet 10 EMFEmma julesNo ratings yet
- Grade 3 Lesson on Changes of MaterialsDocument2 pagesGrade 3 Lesson on Changes of MaterialsJohn Paul M. GalvezNo ratings yet
- Melting of PCM Inside A Novel Encapsulation Design For Thermal EnergyDocument20 pagesMelting of PCM Inside A Novel Encapsulation Design For Thermal EnergyAlirezaNo ratings yet
- CEIC2000 Exam 2016 MainDocument18 pagesCEIC2000 Exam 2016 MainMeena LochniNo ratings yet
- Microsoft Word - 4-State of Matter - Gaseous StateDocument5 pagesMicrosoft Word - 4-State of Matter - Gaseous StateSatya KamNo ratings yet
- Light ScatteringDocument10 pagesLight ScatteringAndri HanryansyahNo ratings yet
- Thermal Properties & Temperature 5 QPDocument9 pagesThermal Properties & Temperature 5 QPJinYoongLimNo ratings yet
- Cycle WorksDocument18 pagesCycle WorksSivarekha KNo ratings yet
- MASS TRANSFER OPERATIONS – II ASSIGNMENT PROBLEMSDocument3 pagesMASS TRANSFER OPERATIONS – II ASSIGNMENT PROBLEMSJAYDEVSINH CHAVDANo ratings yet
- Thermoelectric EffectsDocument43 pagesThermoelectric EffectsLIAKMANNo ratings yet
- HMT Answer 2 & 16 Marks HMTDocument85 pagesHMT Answer 2 & 16 Marks HMTChandra Sekar100% (3)
- Section 4Document3 pagesSection 4Aduchelab AdamsonuniversityNo ratings yet
- Lesson Plan 2Document41 pagesLesson Plan 2Fika Atina RizqianaNo ratings yet
- Toaz - Info Thermodynamics 2 PRDocument6 pagesToaz - Info Thermodynamics 2 PRGerald Kyle A. SupapoNo ratings yet
- How To Select A Compressed Air Dryer - Van Air SystemsDocument3 pagesHow To Select A Compressed Air Dryer - Van Air SystemsHemchand MoreNo ratings yet
- Science 282, 897-901 (1998)Document6 pagesScience 282, 897-901 (1998)Ngọc UyênNo ratings yet
- How To Calculate The Temperature Rise in A Sealed EnclosureDocument20 pagesHow To Calculate The Temperature Rise in A Sealed EnclosureChandrashekhar SarafNo ratings yet
- Spettro Infrarosso Del KCLO3Document6 pagesSpettro Infrarosso Del KCLO3AlessandroNo ratings yet
- 07 GasTreatingDocument52 pages07 GasTreatingHelixNo ratings yet
- Bansal Classes Chemistry Study Material For IIT JEEDocument824 pagesBansal Classes Chemistry Study Material For IIT JEEHarshitShukla0% (1)
- Chapter 13 Reacting Mixtures and CombustionDocument72 pagesChapter 13 Reacting Mixtures and CombustionTimotheus HauwangaNo ratings yet
- 2021-2022 AIChE Student Design Competition Problem StatementDocument28 pages2021-2022 AIChE Student Design Competition Problem StatementUsɱâñ MåâñNo ratings yet
- Air Conditioning (EDITED)Document6 pagesAir Conditioning (EDITED)Justin MercadoNo ratings yet
- 9701 w16 Ms 22Document8 pages9701 w16 Ms 22Praveen PeterNo ratings yet
- Calcium Analysis EDTA TitrationDocument6 pagesCalcium Analysis EDTA TitrationChun Wing Lai100% (2)
- 3.5 Single Mode Laser-External Modulation-Temperature Effort.Document7 pages3.5 Single Mode Laser-External Modulation-Temperature Effort.DEEPAK SNo ratings yet
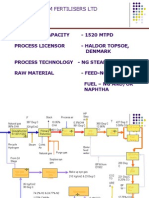
- KRIBHCO SHYAM FERTILISERS LTD AMMONIA PROCESS OVERVIEWDocument51 pagesKRIBHCO SHYAM FERTILISERS LTD AMMONIA PROCESS OVERVIEWSaad Khan89% (9)