You might also like
- GMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsFrom EverandGMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsRating: 5 out of 5 stars5/5 (2)
- The FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsFrom EverandThe FDA and Worldwide Current Good Manufacturing Practices and Quality System Requirements Guidebook for Finished PharmaceuticalsNo ratings yet
- 2010 Article 9503 PDFDocument3 pages2010 Article 9503 PDFlalooprasad15No ratings yet
- ADMET for Medicinal Chemists: A Practical GuideFrom EverandADMET for Medicinal Chemists: A Practical GuideKatya TsaiounNo ratings yet
- 213 1403 1 PBDocument13 pages213 1403 1 PBnahum.bandaNo ratings yet
- How to Integrate Quality by Efficient Design (QbED) in Product DevelopmentFrom EverandHow to Integrate Quality by Efficient Design (QbED) in Product DevelopmentNo ratings yet
- Aksu 2019 Chapter 2Document23 pagesAksu 2019 Chapter 2Angel GarciaNo ratings yet
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionFrom EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNo ratings yet
- Essential Chemistry for Formulators of Semisolid and Liquid DosagesFrom EverandEssential Chemistry for Formulators of Semisolid and Liquid DosagesRating: 5 out of 5 stars5/5 (2)
- Quality by Design: Review ArticleDocument13 pagesQuality by Design: Review ArticleIjupbs IjupbsNo ratings yet
- Some Quality Control Analysis Parameters For Parenteral FormulationsDocument5 pagesSome Quality Control Analysis Parameters For Parenteral FormulationsEditor IJTSRDNo ratings yet
- Nonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsFrom EverandNonclinical Safety Assessment: A Guide to International Pharmaceutical RegulationsWilliam J. BrockNo ratings yet
- Advancement of Emerging Technology Applications To Modernize The Pharmaceutical Manufacturing Base Guidance For IndustryDocument8 pagesAdvancement of Emerging Technology Applications To Modernize The Pharmaceutical Manufacturing Base Guidance For IndustryschumonNo ratings yet
- PAT - A Framework For Innovative Pharmaceutical Development, Manufacturing, and Quality AssuranceDocument19 pagesPAT - A Framework For Innovative Pharmaceutical Development, Manufacturing, and Quality AssuranceHeba El-TayebNo ratings yet
- How to Design and Implement Powder-to-Tablet Continuous Manufacturing SystemsFrom EverandHow to Design and Implement Powder-to-Tablet Continuous Manufacturing SystemsFernando MuzzioNo ratings yet
- Course NotesDocument125 pagesCourse Notesfuji_reihNo ratings yet
- Experiment No. Title Date 1: Definition of Good Pharmacy PracticeDocument457 pagesExperiment No. Title Date 1: Definition of Good Pharmacy Practice10 Adarsh GuptaNo ratings yet
- Regulatory Affairs in Biotech IndustryDocument29 pagesRegulatory Affairs in Biotech IndustryG.Bhavana 4GM20BT018No ratings yet
- Current Good Manufacturing Practices (cGMP) for Pharmaceutical ProductsFrom EverandCurrent Good Manufacturing Practices (cGMP) for Pharmaceutical ProductsNo ratings yet
- Quality Management in PharmaDocument23 pagesQuality Management in PharmaAditi Kaushik100% (1)
- An Overview of Regulatory Affairs in Pharmaceutical IndustryDocument12 pagesAn Overview of Regulatory Affairs in Pharmaceutical Industrypraveen PuttiNo ratings yet
- Quality by Design (QBD) : A Comprehensive Understanding of Implementation and Challenges in Pharmaceuticals DevelopmentDocument8 pagesQuality by Design (QBD) : A Comprehensive Understanding of Implementation and Challenges in Pharmaceuticals DevelopmentmcamilaarredondovelezNo ratings yet
- GMP and Regulations: Good Manufacturing PracticeDocument10 pagesGMP and Regulations: Good Manufacturing PracticeCelia Caroline Neves Faria de SousaNo ratings yet
- TQM in Pharma IndustryDocument23 pagesTQM in Pharma IndustryAditi Kaushik50% (2)
- GMP Requirements For Buildings and Facilities For Api-Comparison of Schedule M, India and Ich Guideline and ApproachDocument33 pagesGMP Requirements For Buildings and Facilities For Api-Comparison of Schedule M, India and Ich Guideline and ApproachManuel AlemanNo ratings yet
- Combination Product (FDC)Document5 pagesCombination Product (FDC)shah777No ratings yet
- Development Knowledge QBDDocument6 pagesDevelopment Knowledge QBDNarendrakumarNo ratings yet
- Good Manufacture PracticeDocument9 pagesGood Manufacture PracticeKateryna BondarenkoNo ratings yet
- Development of Integrated Industrial Process Layout Design For The Production and Quality Control Facility of Various Pharmaceutical FormulationsDocument15 pagesDevelopment of Integrated Industrial Process Layout Design For The Production and Quality Control Facility of Various Pharmaceutical FormulationsAntony Joel Meza LoardoNo ratings yet
- Prodrug Design: Perspectives, Approaches and Applications in Medicinal ChemistryFrom EverandProdrug Design: Perspectives, Approaches and Applications in Medicinal ChemistryRating: 3 out of 5 stars3/5 (1)
- Portfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesFrom EverandPortfolio, Program, and Project Management in the Pharmaceutical and Biotechnology IndustriesPete HarpumNo ratings yet
- GXP Compliance in Pharmaceutical Industry PDFDocument6 pagesGXP Compliance in Pharmaceutical Industry PDFGyana SahooNo ratings yet
- Multivariate Analysis in the Pharmaceutical IndustryFrom EverandMultivariate Analysis in the Pharmaceutical IndustryAna Patricia FerreiraNo ratings yet
- Tirupati MedicareDocument40 pagesTirupati MedicareAmit KumarNo ratings yet
- Biopharmaceutics Modelling As A Fundamental Tool To Support Accelerated AccessDocument20 pagesBiopharmaceutics Modelling As A Fundamental Tool To Support Accelerated AccessMaria Rey RicoNo ratings yet
- Historical Incidents Leading To The Evolution of Good Manufacturing PracticeDocument3 pagesHistorical Incidents Leading To The Evolution of Good Manufacturing PracticeSantosh Kumar TataNo ratings yet
- File-Download Environmental MonitoringDocument8 pagesFile-Download Environmental Monitoringppremala86No ratings yet
- Total Quality Management in Pharmaceuticals PDFDocument11 pagesTotal Quality Management in Pharmaceuticals PDFNarongchai PongpanNo ratings yet
- Quality Systems in The Pharmaceutical IndustryDocument23 pagesQuality Systems in The Pharmaceutical IndustryTimothyPortelliNo ratings yet
- 7.01 CAMs QSE Dec13 v2 1Document48 pages7.01 CAMs QSE Dec13 v2 1dutoitlouwNo ratings yet
- Techniques for Downstream process for Biologic Drugs and VaccinesFrom EverandTechniques for Downstream process for Biologic Drugs and VaccinesNo ratings yet
- JETIR2106052Document18 pagesJETIR2106052ahmunde2002No ratings yet
- 12-15 (1)Document4 pages12-15 (1)Smriti IndoreNo ratings yet
- Good Manufacturing Practice - WKPDocument10 pagesGood Manufacturing Practice - WKPFrancisco Del PuertoNo ratings yet
- Fda NSDDocument27 pagesFda NSDDAVI DE MATOS ALVES alu.ufc.brNo ratings yet
- Ensuring Compliance of Raw Materials and Excipients Across Functional AreasDocument16 pagesEnsuring Compliance of Raw Materials and Excipients Across Functional AreasLinh NguyenNo ratings yet
- Quality by Design (QBD) Approach in Pharmaceuticals: Status, Challenges and Next StepsDocument8 pagesQuality by Design (QBD) Approach in Pharmaceuticals: Status, Challenges and Next StepsnabilahNo ratings yet
- Paper 8873Document10 pagesPaper 8873IJARSCT JournalNo ratings yet
- Quality Assurance and Quality Management in Pharmaceutical IndustryFrom EverandQuality Assurance and Quality Management in Pharmaceutical IndustryRating: 4 out of 5 stars4/5 (4)
- 127 131 (Ijprr)Document5 pages127 131 (Ijprr)Anonymous Dj1jk44kNo ratings yet
- Guidance of Good Manufacturing Practice For Packaging Materials For Medicinal ProductsDocument43 pagesGuidance of Good Manufacturing Practice For Packaging Materials For Medicinal Productshemant singhNo ratings yet
- Pharmacophore Based Drug Design Approach As A Practical Process in Drug DiscoveryDocument13 pagesPharmacophore Based Drug Design Approach As A Practical Process in Drug DiscoverySANJAY K GOWDANo ratings yet
- Quality by Design Approaches For Topical Dermatological Dosage FormsDocument19 pagesQuality by Design Approaches For Topical Dermatological Dosage FormsDoinița UrsuNo ratings yet
- Pharma GMP GuidanceDocument155 pagesPharma GMP GuidanceDinesh SenathipathiNo ratings yet
- Flexo Printing Process ExplainedDocument37 pagesFlexo Printing Process ExplainedHabteyes AbateNo ratings yet
- E-Z WELD PIPE CLEANER SDSDocument2 pagesE-Z WELD PIPE CLEANER SDSVinith KumarNo ratings yet
- 23 M-AstDocument4 pages23 M-AstLegi YangNo ratings yet
- Mtds - Burst 100 RSDDocument2 pagesMtds - Burst 100 RSDDr. Ahmed Abdel-HakimNo ratings yet
- US20030075077A1Document12 pagesUS20030075077A1Kyaw Kyaw LinnNo ratings yet
- Chemistry HL P3Document22 pagesChemistry HL P3Juan Fernando Velasco ForeroNo ratings yet
- Monografia MainDocument20 pagesMonografia MainWILIAM SALVADORNo ratings yet
- IMA- Potentiometric and Conductometric MethodsDocument16 pagesIMA- Potentiometric and Conductometric MethodsUadNo ratings yet
- Swiss Cheese: ALP Science 2008, No. 518Document17 pagesSwiss Cheese: ALP Science 2008, No. 518mostafa el-sammadNo ratings yet
- Jotun Technical Submittal Ver.3Document124 pagesJotun Technical Submittal Ver.3Ahmed AssafNo ratings yet
- General Chemistry 2 q3 Slm4Document15 pagesGeneral Chemistry 2 q3 Slm4Christine Mae LlabanNo ratings yet
- Pozzolanic Reactions of Six Principal Clay Minerals Activaction, Reactivity Assesments and Technological EffectsDocument12 pagesPozzolanic Reactions of Six Principal Clay Minerals Activaction, Reactivity Assesments and Technological EffectsDIAZCORDOBANo ratings yet
- Guggenheim 1935Document57 pagesGuggenheim 1935Niraj ThakreNo ratings yet
- PT - Science 6 - Q1Document7 pagesPT - Science 6 - Q1Airma Ross HernandezNo ratings yet
- AMS-5639 304 - 304L Stainless PDFDocument7 pagesAMS-5639 304 - 304L Stainless PDFJesse KrebsNo ratings yet
- Moores Test and Barfoeds TestDocument3 pagesMoores Test and Barfoeds TestFrancis CaloNo ratings yet
- Determination of PH Value in Mud FluidDocument18 pagesDetermination of PH Value in Mud Fluidn oNo ratings yet
- GAS PROPERTIESDocument2 pagesGAS PROPERTIESsarah thomasNo ratings yet
- Chem 3Document4 pagesChem 3Cristyl Vismanos GastaNo ratings yet
- Pdvsa: Materials Technical SpecificationDocument28 pagesPdvsa: Materials Technical SpecificationYotselys López100% (1)
- Egemaster-Non Sparking Tools Safety PDFDocument5 pagesEgemaster-Non Sparking Tools Safety PDFZulfiqar AliNo ratings yet
- Ionic Liquid-Assisted Refinery Processes - A Review and Industrial PerspectiveDocument23 pagesIonic Liquid-Assisted Refinery Processes - A Review and Industrial Perspectivemohsen miandehiNo ratings yet
- Enzymatic Assay of XYLANASE (EC 3.2.1.8) PrincipleDocument4 pagesEnzymatic Assay of XYLANASE (EC 3.2.1.8) Principlesyaza amiliaNo ratings yet
- Staal 40: Gate ValvesDocument8 pagesStaal 40: Gate ValvesEric LarrondoNo ratings yet
- Year 9 Worksheet on Mixtures and Melting PointsDocument2 pagesYear 9 Worksheet on Mixtures and Melting PointsAlric DsouzaNo ratings yet
- (2023) Extraction of Scandium From Bauxite Residue by High-Pressure (Icp)Document11 pages(2023) Extraction of Scandium From Bauxite Residue by High-Pressure (Icp)Mincen RevaNo ratings yet
- CA 0100 CH 2 X 50 ML CA 0500 CH 4 X 125 ML: LinearityDocument1 pageCA 0100 CH 2 X 50 ML CA 0500 CH 4 X 125 ML: LinearityDharmesh PatelNo ratings yet
- Chemical Reaction - WikipediaDocument10 pagesChemical Reaction - WikipediaMala DeviNo ratings yet
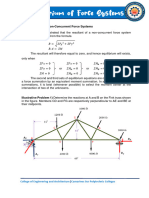
- Chapter 4. Equilibrium of Non Concurrent Force SystemsDocument7 pagesChapter 4. Equilibrium of Non Concurrent Force SystemscarminavegamariaNo ratings yet
- Bmac4E02: Modern Analytical InstrumentationDocument22 pagesBmac4E02: Modern Analytical InstrumentationSachin ashokNo ratings yet