You might also like
- Hematología. Casos clínicos: preguntas y respuestasFrom EverandHematología. Casos clínicos: preguntas y respuestasRating: 4 out of 5 stars4/5 (4)
- GAPS, el síndrome psico-intestinal: Un tratamiento natural para el autismo, la dispraxia, el trastorno por déficit de atención con o sin hiperactividad, la dislexia, la depresión y la esquizofreniaFrom EverandGAPS, el síndrome psico-intestinal: Un tratamiento natural para el autismo, la dispraxia, el trastorno por déficit de atención con o sin hiperactividad, la dislexia, la depresión y la esquizofreniaRating: 3.5 out of 5 stars3.5/5 (12)
- El código del cáncer: Una aproximación nueva y revolucionaria a un misterio médicoFrom EverandEl código del cáncer: Una aproximación nueva y revolucionaria a un misterio médicoRating: 5 out of 5 stars5/5 (6)
- Fundamentos del diagnóstico y tratamiento del cáncer en adultos: Una aproximación inicial para el médico no especialista en cáncerFrom EverandFundamentos del diagnóstico y tratamiento del cáncer en adultos: Una aproximación inicial para el médico no especialista en cáncerNo ratings yet
- Trastorno de Espectro Autista: Una guía con 10 puntos clave para diseñar la estrategia más adecuada para tu hijoFrom EverandTrastorno de Espectro Autista: Una guía con 10 puntos clave para diseñar la estrategia más adecuada para tu hijoNo ratings yet
- Trastorno Por Evitación y o Restricción de La Ingesta de Alimentos (TERIA)Document16 pagesTrastorno Por Evitación y o Restricción de La Ingesta de Alimentos (TERIA)Pepa Pig100% (1)
- Hiperplasia Suprarrenal Congenita: Una Guia Para Los PadresFrom EverandHiperplasia Suprarrenal Congenita: Una Guia Para Los PadresRating: 5 out of 5 stars5/5 (1)
- Mi Sueño Americano: El Viaje De Una Mujer Viviendo Con Una Enfermedad CrónicaFrom EverandMi Sueño Americano: El Viaje De Una Mujer Viviendo Con Una Enfermedad CrónicaNo ratings yet
- Mejorar los cuidados del enfermo dependienteFrom EverandMejorar los cuidados del enfermo dependienteNo ratings yet
- Mejorar los cuidados del enfermo dependienteFrom EverandMejorar los cuidados del enfermo dependienteNo ratings yet
- Bases Anatomopatológicas De La Enfermedad Quirúrgica: Tomo IiFrom EverandBases Anatomopatológicas De La Enfermedad Quirúrgica: Tomo IiNo ratings yet
- Uso del Cannabis en la epilepsia refractaria infantilFrom EverandUso del Cannabis en la epilepsia refractaria infantilNo ratings yet
- Comprendiendo El Alzhéimer: 7 mitos y verdades que debes conocerFrom EverandComprendiendo El Alzhéimer: 7 mitos y verdades que debes conocerRating: 5 out of 5 stars5/5 (1)
- Convivir con el cáncer: Ayuda psicológica para pacientes y familiaresFrom EverandConvivir con el cáncer: Ayuda psicológica para pacientes y familiaresNo ratings yet
- Absorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleFrom EverandAbsorción con filtro BK como terapia eficaz en el riñón del mieloma múltipleNo ratings yet
- La dieta Mind, alimentación que ayuda a prevenir la enfermedad de AlzheimerFrom EverandLa dieta Mind, alimentación que ayuda a prevenir la enfermedad de AlzheimerRating: 3 out of 5 stars3/5 (2)
- Urgencias médicas en el consultorio odontológico: Conocimientos básicos odontológicos, #2From EverandUrgencias médicas en el consultorio odontológico: Conocimientos básicos odontológicos, #2Rating: 5 out of 5 stars5/5 (1)
- GuíaBurros: Cuando tu cuerpo te avisa de la enfermedad: Guía para cuidar tu saludFrom EverandGuíaBurros: Cuando tu cuerpo te avisa de la enfermedad: Guía para cuidar tu saludNo ratings yet
- En defensa propia: Aventuras y desventuras del sistema inmunológicoFrom EverandEn defensa propia: Aventuras y desventuras del sistema inmunológicoRating: 5 out of 5 stars5/5 (1)
- Infectología práctica para el pediatra: Segunda ediciónFrom EverandInfectología práctica para el pediatra: Segunda ediciónRating: 1 out of 5 stars1/5 (1)
- Testimonios De Enfermos Curados De Cáncer Leucemias Linfomas Corazón Y Otras EnfermedadesFrom EverandTestimonios De Enfermos Curados De Cáncer Leucemias Linfomas Corazón Y Otras EnfermedadesNo ratings yet
- El ronquido y la apnea del sueño: Su diagnóstico y tratamiento al alcance de todosFrom EverandEl ronquido y la apnea del sueño: Su diagnóstico y tratamiento al alcance de todosNo ratings yet
- Resumen Y Guía De Estudio – Alimentos Para El CerebroFrom EverandResumen Y Guía De Estudio – Alimentos Para El CerebroRating: 5 out of 5 stars5/5 (1)
- Health, Aging & End of Life. Vol. 6 2021: Revista Internacional de InvestigaciónFrom EverandHealth, Aging & End of Life. Vol. 6 2021: Revista Internacional de InvestigaciónNo ratings yet
- Toxoplasmosis ocular: ¡No coma cuento, ni carne cruda!From EverandToxoplasmosis ocular: ¡No coma cuento, ni carne cruda!No ratings yet
- Tuve Cáncer de mama con invasión a pulmón y ... ¡Estoy Sana!: Experiencia de vida de una guerreraFrom EverandTuve Cáncer de mama con invasión a pulmón y ... ¡Estoy Sana!: Experiencia de vida de una guerreraNo ratings yet
- Resumen Completo - Cerebro De Pan (Grain Brain) - Basado En El Libro De David Perlmutter: (Edicion Extendida)From EverandResumen Completo - Cerebro De Pan (Grain Brain) - Basado En El Libro De David Perlmutter: (Edicion Extendida)No ratings yet
- Para atender bien hay que estar bien: SÍNDROME: Convivir con un enfermo crónicoFrom EverandPara atender bien hay que estar bien: SÍNDROME: Convivir con un enfermo crónicoNo ratings yet
- Resumen Y Guía De Estudio – La Solución Del Alzheimer: Prevenir Y Revertir El Deterioro CognitivoFrom EverandResumen Y Guía De Estudio – La Solución Del Alzheimer: Prevenir Y Revertir El Deterioro CognitivoRating: 5 out of 5 stars5/5 (1)
- Propiedades de los derivados del cannabis en el AlzheimerFrom EverandPropiedades de los derivados del cannabis en el AlzheimerNo ratings yet
- El estudio de China: El estudio más completo jamás realizado sobre nutriciónFrom EverandEl estudio de China: El estudio más completo jamás realizado sobre nutriciónNo ratings yet
- Los fantamas de espejo. Ensayo didáctico: Un compendio de trastornos alimentarios, casuas, consecuencias y solucionesFrom EverandLos fantamas de espejo. Ensayo didáctico: Un compendio de trastornos alimentarios, casuas, consecuencias y solucionesNo ratings yet
- Vivir con Epilepsia Guía Práctica para Pacientes y FamiliaresFrom EverandVivir con Epilepsia Guía Práctica para Pacientes y FamiliaresNo ratings yet
- Niños sanos, adultos sanos: La salud empieza a programarse en el embarazoFrom EverandNiños sanos, adultos sanos: La salud empieza a programarse en el embarazoRating: 4 out of 5 stars4/5 (5)
- Bebé A Bordo: Como curar la infertilidad de forma naturalFrom EverandBebé A Bordo: Como curar la infertilidad de forma naturalRating: 5 out of 5 stars5/5 (1)
- La Lista de Salud: Lo Que Usted y Su Familia Necesita Saber Para Prevenir Enfermedades y Vivir una Vida Larga y SaludableFrom EverandLa Lista de Salud: Lo Que Usted y Su Familia Necesita Saber Para Prevenir Enfermedades y Vivir una Vida Larga y SaludableRating: 4 out of 5 stars4/5 (3)
- Educación alimentaria durante la infancia y el crecimientoFrom EverandEducación alimentaria durante la infancia y el crecimientoNo ratings yet
- ¿Gorditos o enfermos?: La obesidad en niños y adolescentes. Texto para niños y adolescentes, sus papás y sus maestros, para lograr un crecimiento saludableFrom Everand¿Gorditos o enfermos?: La obesidad en niños y adolescentes. Texto para niños y adolescentes, sus papás y sus maestros, para lograr un crecimiento saludableRating: 4 out of 5 stars4/5 (4)
- Nutrición y cancer: Guía para la prevención y tratamiento del cancer (2ª edición)From EverandNutrición y cancer: Guía para la prevención y tratamiento del cancer (2ª edición)Rating: 3.5 out of 5 stars3.5/5 (3)
- Criterios clínicos de enfermedades genéticasFrom EverandCriterios clínicos de enfermedades genéticasRating: 1 out of 5 stars1/5 (1)
- Entendiendo las metabolopatías: Una guía sencilla con ejemplosFrom EverandEntendiendo las metabolopatías: Una guía sencilla con ejemplosNo ratings yet
- Guia Sobre El AstragaloDocument3 pagesGuia Sobre El AstragaloPepa PigNo ratings yet
- Relacion Entre Lo Divino y Dios. EticaDocument14 pagesRelacion Entre Lo Divino y Dios. EticaPepa PigNo ratings yet
- Guia Sobre El LlantenDocument7 pagesGuia Sobre El LlantenPepa PigNo ratings yet
- Guia Sobre La Leucemia FelinaDocument1 pageGuia Sobre La Leucemia FelinaPepa PigNo ratings yet
- TIROIDESDocument356 pagesTIROIDESPepa PigNo ratings yet
- El Papa y El CapitalismoDocument6 pagesEl Papa y El CapitalismoPepa PigNo ratings yet
- Amado Saint Germain - Comunicacion Cosmica - Estudio Sobre Los Siete RayosDocument48 pagesAmado Saint Germain - Comunicacion Cosmica - Estudio Sobre Los Siete RayosPepa PigNo ratings yet
- Los Judíos y Los Asesinatos Ritualísticos de Bebes CristianosDocument69 pagesLos Judíos y Los Asesinatos Ritualísticos de Bebes CristianosPepa PigNo ratings yet
- Simbologia Farmaceutica de Los Origenes de La FarmaciaDocument10 pagesSimbologia Farmaceutica de Los Origenes de La FarmaciaPepa PigNo ratings yet
- Neuroanatomia Del Par CeroDocument6 pagesNeuroanatomia Del Par CeroPepa PigNo ratings yet
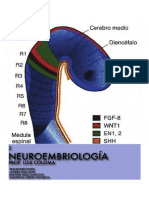
- Embriologia NEURO ApuntesDocument25 pagesEmbriologia NEURO ApuntesPepa PigNo ratings yet
- Rinitis y PsicologiaDocument6 pagesRinitis y PsicologiaPepa PigNo ratings yet
- Diferencias Entre El Policial de Enigma y El NegroDocument2 pagesDiferencias Entre El Policial de Enigma y El NegroPepa PigNo ratings yet
- Fisica - Mecanica de FluidosDocument8 pagesFisica - Mecanica de FluidosPepa PigNo ratings yet
- Las Dimensiones Del Rol Del InspectorDocument19 pagesLas Dimensiones Del Rol Del InspectorPepa PigNo ratings yet
- Las Siete Dimensiones Del Ser - Pablo SenderDocument66 pagesLas Siete Dimensiones Del Ser - Pablo SenderPepa Pig100% (1)
- Apuntes Y Recetas de La Cocina VascaDocument60 pagesApuntes Y Recetas de La Cocina VascaPepa PigNo ratings yet
- Desnivel 419 - Diciembre 2021Document100 pagesDesnivel 419 - Diciembre 2021Pepa PigNo ratings yet
- Hambre, Ingesta de Alimentos y SaludDocument27 pagesHambre, Ingesta de Alimentos y SaludPepa PigNo ratings yet
- Las Dimensiones Del Ser HumanoDocument1 pageLas Dimensiones Del Ser HumanoPepa PigNo ratings yet
- Caso Clinico Sobre El Sindrome CerebelosoDocument5 pagesCaso Clinico Sobre El Sindrome CerebelosoPepa PigNo ratings yet
- CASO CLINICO - Melanoma MetastasicoDocument6 pagesCASO CLINICO - Melanoma MetastasicoPepa PigNo ratings yet
- Lengua - Comentario de Texto Argumentativo - Apreciado LadrónDocument2 pagesLengua - Comentario de Texto Argumentativo - Apreciado LadrónPepa PigNo ratings yet
- El Teletrabajo, El Zoom y La Depresión - Byung-Chul HanDocument6 pagesEl Teletrabajo, El Zoom y La Depresión - Byung-Chul HanPepa PigNo ratings yet
- Introducción A La ArgumentaciónDocument10 pagesIntroducción A La ArgumentaciónPepa PigNo ratings yet
- Introducción A La CartografiaDocument8 pagesIntroducción A La CartografiaPepa PigNo ratings yet
- Argumentacion LiterariaDocument3 pagesArgumentacion LiterariaPepa Pig100% (1)