You might also like
- Freedom Writers Discussion Questions (Complete)Document4 pagesFreedom Writers Discussion Questions (Complete)A_WSM_SIC100% (2)
- Mirko Workout PDFDocument10 pagesMirko Workout PDFSirine Boussama100% (3)
- Hilton Hotema - Man's Higher Consciousness PDFDocument143 pagesHilton Hotema - Man's Higher Consciousness PDFTheLight WorkersMedia92% (12)
- The Sourcebook for Clinical Research: A Practical Guide for Study ConductFrom EverandThe Sourcebook for Clinical Research: A Practical Guide for Study ConductRating: 5 out of 5 stars5/5 (1)
- Language of Clinical TrialsDocument36 pagesLanguage of Clinical Trialsjram00No ratings yet
- Self-Directed Certification Exam Review of GCP For Clinical Research Coordinators, Clinical Research Associates (Monitors) and Clinical InvestigatorsDocument6 pagesSelf-Directed Certification Exam Review of GCP For Clinical Research Coordinators, Clinical Research Associates (Monitors) and Clinical Investigatorsnarmi10No ratings yet
- REGULATORYDocument19 pagesREGULATORYsrishty100% (1)
- FDA Approval GuideDocument38 pagesFDA Approval GuideYuwono WibowoNo ratings yet
- Appendix D: Productivity Evaluation ChecklistDocument16 pagesAppendix D: Productivity Evaluation Checklistabdella.whateverNo ratings yet
- Lavender MsdsDocument7 pagesLavender MsdsRaffandi RolandoNo ratings yet
- IndndaandandaDocument46 pagesIndndaandandaPavani SriramNo ratings yet
- Investigator Responsibilities in Clinical Research Mod 2Document6 pagesInvestigator Responsibilities in Clinical Research Mod 2Raquel VargasNo ratings yet
- 23 - Ensuring The Safety of Clinical Trials-Draft Investigatores & DMC IMPDocument30 pages23 - Ensuring The Safety of Clinical Trials-Draft Investigatores & DMC IMPsightbdNo ratings yet
- CHECKLIST: Post Approval Monitoring - Drug or Device Clinical TrialDocument5 pagesCHECKLIST: Post Approval Monitoring - Drug or Device Clinical TrialAlma EvangelistaNo ratings yet
- Lecture 2 (IND)Document33 pagesLecture 2 (IND)minjiNo ratings yet
- Pediatric GCP FINAL SLeibenhaut - 2.12.19Document65 pagesPediatric GCP FINAL SLeibenhaut - 2.12.19DevangNo ratings yet
- Misha Regulatory AffairsDocument26 pagesMisha Regulatory AffairsGULSHAN MADHURNo ratings yet
- Resources For IND ApplicationDocument19 pagesResources For IND ApplicationVidya RaniNo ratings yet
- Lecture # 8 Dr. Laiq (6.10.19) PDFDocument50 pagesLecture # 8 Dr. Laiq (6.10.19) PDFAbbas HassanNo ratings yet
- CRCP Lecture Reg Approvals Oct 2020Document65 pagesCRCP Lecture Reg Approvals Oct 2020EsEnGauharNo ratings yet
- 11 FDA Perspective On International Clinical Trials - Kassa Ayalew PDFDocument42 pages11 FDA Perspective On International Clinical Trials - Kassa Ayalew PDFVinita KhaidemNo ratings yet
- A Pre-Marketing ActivitiesDocument30 pagesA Pre-Marketing ActivitiesKaye DepabloNo ratings yet
- Schedule YDocument55 pagesSchedule YshilpapillaiNo ratings yet
- Good Clinical PracticesDocument75 pagesGood Clinical PracticesLuisaReyesNo ratings yet
- R&D Group 8 Regulatory Roll No 3,6,11,15,17,40Document49 pagesR&D Group 8 Regulatory Roll No 3,6,11,15,17,40darpan30No ratings yet
- Use of Investigational Drugs or Biologic Products in Human Subjects ResearchDocument6 pagesUse of Investigational Drugs or Biologic Products in Human Subjects ResearchMalik InamNo ratings yet
- Drugs Vs Devices: Is A Clinical Trial Necessary?Document5 pagesDrugs Vs Devices: Is A Clinical Trial Necessary?basakerpolatNo ratings yet
- Ethics CommitteeDocument4 pagesEthics Committeeapi-3810976No ratings yet
- New Drug Application HardDocument37 pagesNew Drug Application HardGANESH KUMAR JELLA100% (1)
- Pharmabizz QADocument15 pagesPharmabizz QArama_v100% (1)
- Use of Investigational Drugs or Biologic Products in Human Subjects Research Policy - Arizona, Florida, RochesterDocument6 pagesUse of Investigational Drugs or Biologic Products in Human Subjects Research Policy - Arizona, Florida, RochesterRajan VermaNo ratings yet
- Seventh Schedule, Medical Device RulesDocument20 pagesSeventh Schedule, Medical Device RulesGurneet Kaur KhalsaNo ratings yet
- Global Regulations in Clinical Trials by N.srinivas ICRIDocument62 pagesGlobal Regulations in Clinical Trials by N.srinivas ICRIravi9247No ratings yet
- Curs GCP: What Is Good Clinical Practice?Document6 pagesCurs GCP: What Is Good Clinical Practice?Delia NituNo ratings yet
- FDA CDX PresentationDocument50 pagesFDA CDX Presentationmartin.dubuc-extNo ratings yet
- Bio-Business in Brief: The Challenges of Clinical TrialsDocument9 pagesBio-Business in Brief: The Challenges of Clinical TrialsDnyanesh LimayeNo ratings yet
- Investigational New Drug Application (INDA)Document25 pagesInvestigational New Drug Application (INDA)Mallikarjun MangapuramNo ratings yet
- Schedule Y New RegulationsDocument55 pagesSchedule Y New RegulationsDinesh PatoleNo ratings yet
- JCG Clinical Trials 2016 V 3.5Document39 pagesJCG Clinical Trials 2016 V 3.5Mohammed HammedNo ratings yet
- INVESTIGATIONALNEWDRUGAPPLICATION INDAaaDocument32 pagesINVESTIGATIONALNEWDRUGAPPLICATION INDAaaHan XuNo ratings yet
- Module 2 - Regulatory Approval Process - Lecture NotesDocument16 pagesModule 2 - Regulatory Approval Process - Lecture NotesyvssmanjunathNo ratings yet
- Bty459 CaDocument9 pagesBty459 Cavivekanand879355443612No ratings yet
- Introduction To TheoryDocument8 pagesIntroduction To TheoryNayan ChaudhariNo ratings yet
- Welcome To Today's FDA/CDRH WebinarDocument35 pagesWelcome To Today's FDA/CDRH WebinarXunyao LuNo ratings yet
- 9 - DR Arun Bhatt - Schedule YDocument27 pages9 - DR Arun Bhatt - Schedule Yvivek100% (1)
- Global Submission On IndDocument16 pagesGlobal Submission On IndRahul PalsNo ratings yet
- FDA Inspections of Clinical Investigators - Information Sheet PDFDocument9 pagesFDA Inspections of Clinical Investigators - Information Sheet PDFJudit SzepesiNo ratings yet
- 1556-FDA Audit Preparation Checklist - NAV - 03JAN13Document26 pages1556-FDA Audit Preparation Checklist - NAV - 03JAN13Sandeep Somaiya100% (1)
- PV Regulatory Affairs Ut 2.1Document18 pagesPV Regulatory Affairs Ut 2.150KMKDIVYA RAJPALNo ratings yet
- Drug Approval Process 12 Feb-2014 Sandeep BatraDocument47 pagesDrug Approval Process 12 Feb-2014 Sandeep BatraSandeep BatraNo ratings yet
- Drug Development Process Cleveland, 6.23.06Document65 pagesDrug Development Process Cleveland, 6.23.06Srinivasa Chary SriramadasuNo ratings yet
- Konsep GCP Good Clinical PracticeDocument48 pagesKonsep GCP Good Clinical PracticeeuhsahaNo ratings yet
- MTN - IoR Training - Slides Notes - FINAL - 06.30.16Document64 pagesMTN - IoR Training - Slides Notes - FINAL - 06.30.16luamsmarinsNo ratings yet
- DRA Unit IIDocument134 pagesDRA Unit IIOyshi RaoNo ratings yet
- DCS CR Final Exam Question FileDocument23 pagesDCS CR Final Exam Question FileAnish RedkarNo ratings yet
- HC Inspection Process Guidance Document Final Dec 15 2015 3Document25 pagesHC Inspection Process Guidance Document Final Dec 15 2015 3Dawn CasuncadNo ratings yet
- Summary PDFDocument3 pagesSummary PDFHari ThekkethilNo ratings yet
- 1 Indndaandasnda-210220130515Document21 pages1 Indndaandasnda-210220130515DeepaNo ratings yet
- Investigational New Drug Application (IND)Document48 pagesInvestigational New Drug Application (IND)Divya100% (1)
- Ind Studies by Sejal KhumanDocument34 pagesInd Studies by Sejal KhumanSejal khumamNo ratings yet
- Walker CanadianandInternationalStandardsDocument43 pagesWalker CanadianandInternationalStandardsRenzoCostaNo ratings yet
- Investigational DrugsDocument34 pagesInvestigational DrugsIVORY DIANE AMANCIONo ratings yet
- Expanded Access Clinical TrialDocument134 pagesExpanded Access Clinical TrialsiddharthkamerkarNo ratings yet
- Code of Federal Regulations Title 45 - Public Welfare Department of Health and Human Services Protection of Human SubjectsDocument12 pagesCode of Federal Regulations Title 45 - Public Welfare Department of Health and Human Services Protection of Human SubjectsthebigfluffyNo ratings yet
- Good Clinical Practice Guidelines IndiaDocument4 pagesGood Clinical Practice Guidelines IndiaMonica0% (1)
- Follow The Path To UCD - Offer Holders Next StepsDocument19 pagesFollow The Path To UCD - Offer Holders Next StepsManna PintoNo ratings yet
- Iontophoresis:: Iontophoresis Along With Various Drugs UsedDocument6 pagesIontophoresis:: Iontophoresis Along With Various Drugs UsedApoorvNo ratings yet
- Futuristic NursingDocument15 pagesFuturistic Nursingmanu sethi79% (19)
- Case - History PsychiatryDocument28 pagesCase - History PsychiatryMahmood SalahNo ratings yet
- Life Skill Development Solved MCQs (Set-4)Document8 pagesLife Skill Development Solved MCQs (Set-4)funny worldNo ratings yet
- Silver Book 5th Edn Part A DeprescribingDocument6 pagesSilver Book 5th Edn Part A DeprescribingAnton BalansagNo ratings yet
- Kala BakraDocument2 pagesKala BakraIrfanNo ratings yet
- 01.work Instruction For Operation of Belt Conveyor-UpdatedDocument6 pages01.work Instruction For Operation of Belt Conveyor-UpdatedRahul VermaNo ratings yet
- Assay SolutionsDocument1 pageAssay SolutionsAlex RamirezNo ratings yet
- Week 2 Rle ActivityDocument3 pagesWeek 2 Rle ActivityMICHELLE BIANCA PATRICE CRUZNo ratings yet
- Botanical Safety Assessment GuidelinesDocument11 pagesBotanical Safety Assessment GuidelinesRaida SiagianNo ratings yet
- Heart Failure and Stroke Risks in Users of Liothyronine + LT4 Vs Lt4 Alone 2022Document8 pagesHeart Failure and Stroke Risks in Users of Liothyronine + LT4 Vs Lt4 Alone 2022NAYSHA YANET CHAVEZ RONDINELNo ratings yet
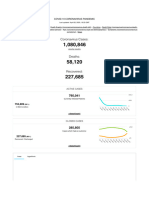
- Coronavirus Update (Live) : 1,080,846 Cases and 58,120 Deaths From COVID-19 Virus Outbreak - WorldometerDocument20 pagesCoronavirus Update (Live) : 1,080,846 Cases and 58,120 Deaths From COVID-19 Virus Outbreak - Worldometernilton lialungaNo ratings yet
- Traditional Assessment Vs Authentic AssessmentDocument4 pagesTraditional Assessment Vs Authentic AssessmentAngelo LagmayNo ratings yet
- Unit-2 (Part-1) Nutraceuticals: B. Pharm 6 SemDocument49 pagesUnit-2 (Part-1) Nutraceuticals: B. Pharm 6 SemMamta Pant75% (4)
- Letter of AccrualDocument2 pagesLetter of Accrualmodi21xyzNo ratings yet
- Student Worksheet - How We Get Our Skin ColorDocument8 pagesStudent Worksheet - How We Get Our Skin ColorAndreu Angeles20% (1)
- S22a PDFDocument32 pagesS22a PDFjuanNo ratings yet
- Cit Mikrotalasna Rezonantna Terapija Patent STR 1Document10 pagesCit Mikrotalasna Rezonantna Terapija Patent STR 1Jovo Kurtovic JojaNo ratings yet
- NASKAH SOAL UM Bhs InggrisDocument10 pagesNASKAH SOAL UM Bhs InggrisSetiawan ArulNo ratings yet
- Annex D Health Declaretion FormDocument2 pagesAnnex D Health Declaretion FormJoseph SadiaNo ratings yet
- Sobre El Amor - Carl Gustav JungDocument51 pagesSobre El Amor - Carl Gustav JungPaula Noelia BenítezNo ratings yet
- Resource Unit For Ward Class TopicsDocument3 pagesResource Unit For Ward Class TopicsMonica JubaneNo ratings yet
- Positive Education: An Introduction: October 2015Document5 pagesPositive Education: An Introduction: October 2015Danielle GraceNo ratings yet
- S/5 ADU Carestation: An Integrated Solution For Quality CareDocument6 pagesS/5 ADU Carestation: An Integrated Solution For Quality CareJuan AmaroNo ratings yet