You might also like
- Translational Research in Coronary Artery Disease: Pathophysiology to TreatmentFrom EverandTranslational Research in Coronary Artery Disease: Pathophysiology to TreatmentNo ratings yet
- New Progress in Diagnosis and Treatment of Pulmonary Arterial HypertensionDocument9 pagesNew Progress in Diagnosis and Treatment of Pulmonary Arterial HypertensionRhahahaNo ratings yet
- Clinical Updates in the Management of Severe Asthma: New Strategies for Individualizing Long-term CareFrom EverandClinical Updates in the Management of Severe Asthma: New Strategies for Individualizing Long-term CareNo ratings yet
- Ijms 24 08462Document4 pagesIjms 24 08462RhahahaNo ratings yet
- Insights On The Epigenetic MechanismsDocument12 pagesInsights On The Epigenetic MechanismsrafaellapordeusNo ratings yet
- Secondary HypertensionFrom EverandSecondary HypertensionAlberto MorgantiNo ratings yet
- Metabolic Dysfunction in Pulmonary Hypertension FR PDFDocument11 pagesMetabolic Dysfunction in Pulmonary Hypertension FR PDFWayan GunawanNo ratings yet
- Genetic Pathway PAHDocument9 pagesGenetic Pathway PAHafifahNo ratings yet
- SCUBE1 Controls BMPR2-Relevant Pulmonary Endothelial FunctionDocument20 pagesSCUBE1 Controls BMPR2-Relevant Pulmonary Endothelial FunctionLeidy LambertinezNo ratings yet
- CRP2020 7526508Document10 pagesCRP2020 7526508Faradiba MaricarNo ratings yet
- Effect of Beraprost On Pulmonary Hypertension DueDocument6 pagesEffect of Beraprost On Pulmonary Hypertension DueBabebos 95No ratings yet
- Natural Ampk ActivatorsDocument15 pagesNatural Ampk ActivatorsSubbaraju Gv100% (1)
- British J Pharmacology - 2021 - DignamDocument27 pagesBritish J Pharmacology - 2021 - DignamLinda Silvana SariNo ratings yet
- Pharmaceuticals 15 01242Document29 pagesPharmaceuticals 15 01242gadangNo ratings yet
- Novel Targets of Drug Treatment For Pulmonary Hypertension: Key PointsDocument10 pagesNovel Targets of Drug Treatment For Pulmonary Hypertension: Key PointsgpatwallahNo ratings yet
- Reference Interval For Immature Platelet Fraction On Sysmex XN HaematologyDocument6 pagesReference Interval For Immature Platelet Fraction On Sysmex XN HaematologyTuan NguyenNo ratings yet
- Artculo 2Document8 pagesArtculo 2Estefany CarrascalNo ratings yet
- Advanced Proteomics and Cluster Analysis For Identifying Novel Obstructive Sleep Apnea Subtypes Before and After CPAP TherapyDocument34 pagesAdvanced Proteomics and Cluster Analysis For Identifying Novel Obstructive Sleep Apnea Subtypes Before and After CPAP Therapy马三强No ratings yet
- 1 s2.0 S0954611120302389 MainDocument6 pages1 s2.0 S0954611120302389 Mainlidi booksNo ratings yet
- Lan Nan 2014Document6 pagesLan Nan 2014Juanin Arevalo CifuentesNo ratings yet
- Inflamasi Dan HAPDocument10 pagesInflamasi Dan HAPAudylia HartonoNo ratings yet
- Nihms 971496Document23 pagesNihms 971496prk prkNo ratings yet
- 10.1016/j.vph.2015.08.005: Vascular PharmacologyDocument24 pages10.1016/j.vph.2015.08.005: Vascular PharmacologyErdelyi-Molnár ImolaNo ratings yet
- Ejhf 2664Document16 pagesEjhf 2664GdfgdFdfdfNo ratings yet
- J Jcin 2022 10 004Document3 pagesJ Jcin 2022 10 004Ivan BitunjacNo ratings yet
- NIH Public AccessDocument18 pagesNIH Public AccessSofia BottiNo ratings yet
- European J of Heart Fail - 2022 - N Ez - Congestion in Heart Failure A Circulating Biomarker Based Perspective A ReviewDocument16 pagesEuropean J of Heart Fail - 2022 - N Ez - Congestion in Heart Failure A Circulating Biomarker Based Perspective A ReviewsilviaNo ratings yet
- PLT Indices Normal ValuesDocument6 pagesPLT Indices Normal ValueskireetiNo ratings yet
- Etm 07 03 0604Document5 pagesEtm 07 03 0604Raissa Metasari TantoNo ratings yet
- Mutational Analysis and Genotype-Phenotype Relation in FHDocument6 pagesMutational Analysis and Genotype-Phenotype Relation in FHChris ChrisNo ratings yet
- 1 s2.0 S0422763817300055 MainDocument20 pages1 s2.0 S0422763817300055 MainNicoleta Popa-FoteaNo ratings yet
- The Prognostic Role of The Spleen in Heart FailureDocument11 pagesThe Prognostic Role of The Spleen in Heart FailurePedro MatosNo ratings yet
- Acc 2021 00927Document9 pagesAcc 2021 00927DON TVNo ratings yet
- Uel and 2015Document7 pagesUel and 2015Nicoleta Popa-FoteaNo ratings yet
- Vinicio de Jesus Perez PDFDocument9 pagesVinicio de Jesus Perez PDFangelicaNo ratings yet
- Meeting-Report-From-The-2nd-International-Symposium-On-New-Frontiers-In-Cardiovascular-Research-Protecting-The-Cardiovascular-System-From-Ischemia-Between-Bench-And-Bedside 2Document13 pagesMeeting-Report-From-The-2nd-International-Symposium-On-New-Frontiers-In-Cardiovascular-Research-Protecting-The-Cardiovascular-System-From-Ischemia-Between-Bench-And-Bedside 2Shreyansh JainNo ratings yet
- 1 s2.0 S1053077023009060 MainDocument31 pages1 s2.0 S1053077023009060 MainedwardmenesesNo ratings yet
- Pathophysiology of COPDDocument4 pagesPathophysiology of COPDrinieeeNo ratings yet
- Proteome Profiling of Lung Tissues in ChronicDocument8 pagesProteome Profiling of Lung Tissues in ChronicAndreea MoalesNo ratings yet
- BBA ClinicalDocument7 pagesBBA ClinicalAyus diningsihNo ratings yet
- CDK 019 Hipertensi & KardiovaskulerDocument55 pagesCDK 019 Hipertensi & Kardiovaskulerrevliee100% (3)
- Clinical Cardiology - 2022 - Liang - Prognostic Value of RDW Alone and in Combination With NT proBNP in Patients With HeartDocument13 pagesClinical Cardiology - 2022 - Liang - Prognostic Value of RDW Alone and in Combination With NT proBNP in Patients With Heartrizkiyah prabawantiNo ratings yet
- Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension - An Immunological PerspectiveDocument23 pagesPulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension - An Immunological PerspectiveHoracioNo ratings yet
- Prognostic Factors in Pulmonary Arterial Hypertension: Literature ReviewDocument5 pagesPrognostic Factors in Pulmonary Arterial Hypertension: Literature Reviewhuda riyambodoNo ratings yet
- European J of Heart Fail 2022 Núñez Congestion in Heart FailureDocument16 pagesEuropean J of Heart Fail 2022 Núñez Congestion in Heart Failurejhoel cruzNo ratings yet
- Endo 2012Document7 pagesEndo 2012Muhammad Anshory AkNo ratings yet
- RCCM 202012-4317soDocument16 pagesRCCM 202012-4317soRhahahaNo ratings yet
- Jama 2022 327 1379Document13 pagesJama 2022 327 1379silviaNo ratings yet
- Frequency and Predictors of Pulmonary Hypertension in Patients With Systemic Lupus ErythematosusDocument4 pagesFrequency and Predictors of Pulmonary Hypertension in Patients With Systemic Lupus ErythematosusAndi WahyudiNo ratings yet
- 1 s2.0 S0022347616310605Document7 pages1 s2.0 S0022347616310605Adrian KhomanNo ratings yet
- Literature Review Acute Kidney InjuryDocument4 pagesLiterature Review Acute Kidney Injuryea813c29100% (1)
- Clinical Relevance of High Sensitivity C-Reactive Protein in CardiologyDocument10 pagesClinical Relevance of High Sensitivity C-Reactive Protein in CardiologybayuwinotoNo ratings yet
- Ijms 20 03991Document7 pagesIjms 20 03991stevenburrow06No ratings yet
- CVS Toxicity Anticancer Systematic ReviewDocument15 pagesCVS Toxicity Anticancer Systematic ReviewprasanbhandariNo ratings yet
- Diagnosis and Treatment of Pulmonary Arterial Hypertension A ReviewDocument13 pagesDiagnosis and Treatment of Pulmonary Arterial Hypertension A ReviewLuis Enrique Giraldo PenicheNo ratings yet
- 1 s2.0 S000287032200093X MainDocument11 pages1 s2.0 S000287032200093X Mainwoldekidansamuel299No ratings yet
- Biomarkers in Heart FailureDocument6 pagesBiomarkers in Heart Failurekevin ortegaNo ratings yet
- Cardiovascular Events Associated With Rofecoxib: Fi Nal Analysis of The Approve TrialDocument9 pagesCardiovascular Events Associated With Rofecoxib: Fi Nal Analysis of The Approve TrialYariCárdenasNo ratings yet
- Biomarker Discovery in Cardio-Oncology.Document8 pagesBiomarker Discovery in Cardio-Oncology.Ștefan SpînuNo ratings yet
- Adrenergic Agonists 2020 PDFDocument65 pagesAdrenergic Agonists 2020 PDFAlaa NaserNo ratings yet
- Siwes ReportDocument23 pagesSiwes ReportIbrahim BadmusNo ratings yet
- CHN Lec - Unit 1 and 2Document4 pagesCHN Lec - Unit 1 and 2Bryan MedranoNo ratings yet
- Alendronate SodiumDocument16 pagesAlendronate SodiumasdwasdNo ratings yet
- Legg-Calve-Perthes Disease Current ConceptsDocument10 pagesLegg-Calve-Perthes Disease Current Conceptsyarimar hoyosNo ratings yet
- The Effect of Tobacco Smoking Among Third Year Student Nurse in The University of LuzonDocument6 pagesThe Effect of Tobacco Smoking Among Third Year Student Nurse in The University of LuzonNeil Christian TadzNo ratings yet
- Índice de ROX Covid 19Document9 pagesÍndice de ROX Covid 19Alejandro LucioNo ratings yet
- Updated - Syllabus MBP 108 (Micro)Document15 pagesUpdated - Syllabus MBP 108 (Micro)Angelo CruzNo ratings yet
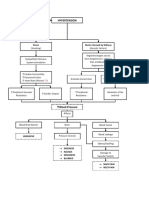
- Hypertension Concept MapDocument1 pageHypertension Concept Mapjyd parreñoNo ratings yet
- Vortioxetine On Cognition in Schizophrenia: Materials and Methods Study DesignDocument5 pagesVortioxetine On Cognition in Schizophrenia: Materials and Methods Study Designhevi_tarsumNo ratings yet
- Mortuary ServicesDocument30 pagesMortuary ServicesYogesh ChandraNo ratings yet
- Heart Rate Variability Threshold As An Alternative.25Document6 pagesHeart Rate Variability Threshold As An Alternative.25Wasly SilvaNo ratings yet
- CDC Interim Reopening GuidanceDocument62 pagesCDC Interim Reopening GuidanceAlex GeliNo ratings yet
- CoronavirusDocument3 pagesCoronavirusĂhméđ ĂĺãshqãrNo ratings yet
- 2023 Herrera Et Al - EFP Guidelines Prevention and Treatment of Peri-Implant DiseasesDocument73 pages2023 Herrera Et Al - EFP Guidelines Prevention and Treatment of Peri-Implant DiseasesGustavo Avila OrtizNo ratings yet
- Bangladesh MBBS Curriculum 2012Document489 pagesBangladesh MBBS Curriculum 2012mail_rajib100% (1)
- Physical DisabilitiesDocument11 pagesPhysical DisabilitiesBobo Wong100% (2)
- Pressure UlcerDocument16 pagesPressure UlcerEloisaNo ratings yet
- LIFOTRONICDocument2 pagesLIFOTRONICPameluska OcampoNo ratings yet
- Acute Leukemia: Thirunavukkarasu MurugappanDocument22 pagesAcute Leukemia: Thirunavukkarasu MurugappanFelix Allen100% (1)
- JPM 12 01580 - 2022Document10 pagesJPM 12 01580 - 2022mihail.boldeanuNo ratings yet
- Sydenham's ChoreaDocument11 pagesSydenham's ChoreaKosi UdohNo ratings yet
- Sjogren's Syndrome - Trishna Predominant Amavata ? A Case ReportDocument5 pagesSjogren's Syndrome - Trishna Predominant Amavata ? A Case ReportSofía MartínezNo ratings yet
- Guidline of Management of Porta CathDocument8 pagesGuidline of Management of Porta CathroncekeyNo ratings yet
- Clinical BacteriologyDocument123 pagesClinical Bacteriologykurotsukki todoroki100% (1)
- Nurse Patient RelationshipDocument29 pagesNurse Patient Relationshipaseel jamalNo ratings yet
- Constitutionalist: Asa My Concern Is The True Meaning and Application of The ConstitutionDocument15 pagesConstitutionalist: Asa My Concern Is The True Meaning and Application of The ConstitutionGerrit Hendrik Schorel-HlavkaNo ratings yet
- BRONCHOPULMONARY DYSPLASIA ModifiedDocument48 pagesBRONCHOPULMONARY DYSPLASIA ModifiedajanmjNo ratings yet
- ER CASE CONFERENCE - Melena 8 - 31 - 21Document33 pagesER CASE CONFERENCE - Melena 8 - 31 - 21Jolaine ValloNo ratings yet
- AU-Strategi Nasional Mutu Pelayanan Kes-Nov 2019Document92 pagesAU-Strategi Nasional Mutu Pelayanan Kes-Nov 2019Ika Belia PratiwiNo ratings yet
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDFrom EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDRating: 5 out of 5 stars5/5 (5)
- LIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionFrom EverandLIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionRating: 4 out of 5 stars4/5 (404)
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)From EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Rating: 3 out of 5 stars3/5 (1)
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedRating: 4.5 out of 5 stars4.5/5 (84)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.From EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Rating: 4.5 out of 5 stars4.5/5 (110)
- The Twentysomething Treatment: A Revolutionary Remedy for an Uncertain AgeFrom EverandThe Twentysomething Treatment: A Revolutionary Remedy for an Uncertain AgeRating: 5 out of 5 stars5/5 (4)
- The Age of Magical Overthinking: Notes on Modern IrrationalityFrom EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityRating: 4 out of 5 stars4/5 (39)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsFrom EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsNo ratings yet
- The Obesity Code: Unlocking the Secrets of Weight LossFrom EverandThe Obesity Code: Unlocking the Secrets of Weight LossRating: 4 out of 5 stars4/5 (6)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaFrom EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaRating: 4.5 out of 5 stars4.5/5 (267)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsFrom EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsRating: 5 out of 5 stars5/5 (1)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsFrom EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsRating: 4 out of 5 stars4/5 (5)
- I Shouldn't Feel This Way: Name What’s Hard, Tame Your Guilt, and Transform Self-Sabotage into Brave ActionFrom EverandI Shouldn't Feel This Way: Name What’s Hard, Tame Your Guilt, and Transform Self-Sabotage into Brave ActionNo ratings yet
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisFrom EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisRating: 4.5 out of 5 stars4.5/5 (44)
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeFrom EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeRating: 2 out of 5 stars2/5 (1)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsFrom EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsRating: 4.5 out of 5 stars4.5/5 (170)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryFrom EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryRating: 4 out of 5 stars4/5 (46)
- Summary: It Didn't Start with You: How Inherited Family Trauma Shapes Who We Are and How to End the Cycle By Mark Wolynn: Key Takeaways, Summary & AnalysisFrom EverandSummary: It Didn't Start with You: How Inherited Family Trauma Shapes Who We Are and How to End the Cycle By Mark Wolynn: Key Takeaways, Summary & AnalysisRating: 5 out of 5 stars5/5 (4)
- To Explain the World: The Discovery of Modern ScienceFrom EverandTo Explain the World: The Discovery of Modern ScienceRating: 3.5 out of 5 stars3.5/5 (51)
- Manipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesFrom EverandManipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesRating: 4.5 out of 5 stars4.5/5 (1412)
- Self-Care for Autistic People: 100+ Ways to Recharge, De-Stress, and Unmask!From EverandSelf-Care for Autistic People: 100+ Ways to Recharge, De-Stress, and Unmask!Rating: 5 out of 5 stars5/5 (1)