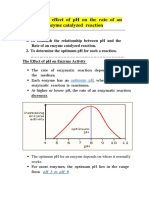
You might also like
- RMIT Design Hub Case Study PDFDocument4 pagesRMIT Design Hub Case Study PDFPavani MalajiNo ratings yet
- Extraction and Characterization of ProteinsDocument4 pagesExtraction and Characterization of ProteinsDozdi100% (2)
- Co- and Post-Translational Modifications of Therapeutic Antibodies and ProteinsFrom EverandCo- and Post-Translational Modifications of Therapeutic Antibodies and ProteinsNo ratings yet
- Bgas Question and AnswerDocument25 pagesBgas Question and AnswerMohammad Aamir Perwaiz94% (16)
- CSWIP 3.1 Multichoice With AnswerDocument18 pagesCSWIP 3.1 Multichoice With AnswerBassamOmarFarghl100% (2)
- Lab 9 - 022-023Document4 pagesLab 9 - 022-023Noor JanahiNo ratings yet
- HBTBC 212 PractDocument8 pagesHBTBC 212 Practtakawira chirimeNo ratings yet
- Protein Membrane Overlay AssayDocument4 pagesProtein Membrane Overlay AssayZain BaderNo ratings yet
- NHS Sulfo-NHS: InstructionsDocument4 pagesNHS Sulfo-NHS: Instructionssantosh_dengaleNo ratings yet
- Determination of Protein Content SpectrophotometricallyDocument10 pagesDetermination of Protein Content SpectrophotometricallyTsabit AlbananiNo ratings yet
- Analysis of Wheat Germ Acid Phosphatase Lab Week 6Document5 pagesAnalysis of Wheat Germ Acid Phosphatase Lab Week 6Jay PatelNo ratings yet
- Answer Questions02 03Document7 pagesAnswer Questions02 03yo-chengNo ratings yet
- Laboratory Report Pharmaceutical Biotechnology: National University of Ho Chi Minh City International UniversityDocument19 pagesLaboratory Report Pharmaceutical Biotechnology: National University of Ho Chi Minh City International UniversityAn Ngoc HoaiNo ratings yet
- AbstractDocument17 pagesAbstractAdrian JayadiNo ratings yet
- MBB 343 Lab, Experiment I Recombinant Plasmid and Protein Expression Outline Week 1Document33 pagesMBB 343 Lab, Experiment I Recombinant Plasmid and Protein Expression Outline Week 1Lilly WilsonNo ratings yet
- Natural Proteins - Sources, Isolation, Characterization and ApplicationsDocument7 pagesNatural Proteins - Sources, Isolation, Characterization and ApplicationsУдшфиАгутеуыNo ratings yet
- Succinate Dehydrogenase Activity of MitochondriaDocument7 pagesSuccinate Dehydrogenase Activity of MitochondriaSherlock Wesley ConanNo ratings yet
- A Simplification of The Protein Assay Method of Lowry Et Al. Which Is More Generally Applicable PDFDocument11 pagesA Simplification of The Protein Assay Method of Lowry Et Al. Which Is More Generally Applicable PDFTania SandovalNo ratings yet
- Enzyme Activity 04Document5 pagesEnzyme Activity 04Dhandapani PerumalNo ratings yet
- Sterol Carrier Protein and Fatty Acid-Binding ProteinDocument7 pagesSterol Carrier Protein and Fatty Acid-Binding ProteinMartin OreNo ratings yet
- MBG312 Chp23 BDDocument47 pagesMBG312 Chp23 BDBaran KirdarNo ratings yet
- Extraction of Native Protein From YeastDocument5 pagesExtraction of Native Protein From YeastBrandon Lam100% (1)
- Protein AssayDocument2 pagesProtein AssayLavinia MihaiNo ratings yet
- Modified Lowry Protein AssayDocument6 pagesModified Lowry Protein AssaywakeyNo ratings yet
- Exercise 5.1 & 5.2 Techniques For Protein Analysis Protein Isolation & Determination of Protein Concentration by SpectrophotometryDocument7 pagesExercise 5.1 & 5.2 Techniques For Protein Analysis Protein Isolation & Determination of Protein Concentration by SpectrophotometryJulie Ann Estaras FelicesNo ratings yet
- Ion Exchage ChromatographyDocument6 pagesIon Exchage ChromatographyRavish Rana100% (2)
- Different Assays To Detect Presence of Activity of Enzymes in Specific OrganelleDocument8 pagesDifferent Assays To Detect Presence of Activity of Enzymes in Specific Organellefrancis harvey rodulfo100% (1)
- Cell Viability Assays Based On Mithochondrial Enzyme PresentationDocument21 pagesCell Viability Assays Based On Mithochondrial Enzyme PresentationmustafabkrlnxNo ratings yet
- Quick Start Bradford Protein Assay: Instruction ManualDocument36 pagesQuick Start Bradford Protein Assay: Instruction ManualTanmoy Kumar DeyNo ratings yet
- Penentuan Kadar Protein Dalam Albumin Telur Melalui Metode LowryDocument7 pagesPenentuan Kadar Protein Dalam Albumin Telur Melalui Metode LowryKadek Anggra SupraptaNo ratings yet
- Biochemistry Problem SetDocument69 pagesBiochemistry Problem Setorion57854375100% (5)
- Protein 2Document12 pagesProtein 2Veysi KızmazNo ratings yet
- C5 Plasmid Isolation Formal ReportDocument9 pagesC5 Plasmid Isolation Formal ReportTanisha ChowdharyNo ratings yet
- CH 5 Lecture SlidesDocument83 pagesCH 5 Lecture SlidesUyên Trần NhưNo ratings yet
- CH 5 Ky Thua Protein-Môn hóa sinh họcDocument83 pagesCH 5 Ky Thua Protein-Môn hóa sinh họcNam NguyenHoangNo ratings yet
- DSP Lab ManualDocument27 pagesDSP Lab ManualRaghav SureshNo ratings yet
- Cell Disruption by Chemical or Enzymatic MethodDocument9 pagesCell Disruption by Chemical or Enzymatic Methodbiovijay101100% (1)
- Biochemistry Final Review: Hartree Lowry Method To Determine Soluble Protein in A SampleDocument18 pagesBiochemistry Final Review: Hartree Lowry Method To Determine Soluble Protein in A SampleLê ThắngNo ratings yet
- Indian Institute of Technology, Bombay. Lecture-10. Proetein-Small Molecule Interaction Study: Immobilization and Binding AnalysisDocument24 pagesIndian Institute of Technology, Bombay. Lecture-10. Proetein-Small Molecule Interaction Study: Immobilization and Binding AnalysisAl AmeenNo ratings yet
- 2021 12 05 471285v2 FullDocument27 pages2021 12 05 471285v2 Fullfatinnafis27No ratings yet
- Ehsan Real IADocument3 pagesEhsan Real IAEhsan MusafirNo ratings yet
- Weber K. y Osborn M. 1969. The Reliability of Molecular Weight Determinations by DodecylDocument7 pagesWeber K. y Osborn M. 1969. The Reliability of Molecular Weight Determinations by DodecylEsaú Bojorquez VelazquezNo ratings yet
- Cell MolecreportDocument8 pagesCell Molecreportapi-586722985No ratings yet
- Problem SetDocument69 pagesProblem SetchemggNo ratings yet
- Protein Isolation From Whole BloodDocument8 pagesProtein Isolation From Whole BloodShadia HeyariNo ratings yet
- Isolation of Invertase PHDocument4 pagesIsolation of Invertase PHJohn Mark Flores VillenaNo ratings yet
- BIO 103 Lab 4 Bradford AssayDocument4 pagesBIO 103 Lab 4 Bradford AssayMaheshMeenaNo ratings yet
- Proteins: Laboratory Manual For Practical Exercises ProteinsDocument5 pagesProteins: Laboratory Manual For Practical Exercises ProteinsSaraNo ratings yet
- Protein Chromatography: Ion Exchange Chromatography AimsDocument4 pagesProtein Chromatography: Ion Exchange Chromatography AimskapilphysioNo ratings yet
- Salting IN, Salting OUT, and Dialysis of Proteins.: ObjectiveDocument8 pagesSalting IN, Salting OUT, and Dialysis of Proteins.: ObjectiveIsla CalugayNo ratings yet
- Gel ElectrophoresisDocument5 pagesGel ElectrophoresisROMELIE CHELSEA ESCOBIANo ratings yet
- The Biosynthesis and Primary Structure of Pea Seed LectinDocument6 pagesThe Biosynthesis and Primary Structure of Pea Seed LectinDelphiNo ratings yet
- Robin Solanki Botony Assignment 2Document6 pagesRobin Solanki Botony Assignment 2Saloni ChaudharyNo ratings yet
- Lab 10 ElectrophoresisDocument4 pagesLab 10 ElectrophoresisErica Sujung ChoeNo ratings yet
- Effect of PH On Enzyme ActivityDocument12 pagesEffect of PH On Enzyme ActivityAb AbNo ratings yet
- ELx808 Determining Total Protein Lowry Method PDFDocument4 pagesELx808 Determining Total Protein Lowry Method PDFAnju GuptaNo ratings yet
- PTM by SamsonDocument28 pagesPTM by SamsonSathish KumarNo ratings yet
- Amino AcidsDocument35 pagesAmino AcidsMd. Muhaiminul IslamNo ratings yet
- Full Lab Report On: Exercise No. 4 Protein DenaturationDocument8 pagesFull Lab Report On: Exercise No. 4 Protein DenaturationElaine FaloNo ratings yet
- Enzyme Kinetics - Lab Manual 02-16-09 HMDocument10 pagesEnzyme Kinetics - Lab Manual 02-16-09 HMFoudiKarimNo ratings yet
- Lab Demo Lowrys MethodDocument20 pagesLab Demo Lowrys MethodM. GokulakannanNo ratings yet
- 9-10 Protein & Analisis ProteinDocument52 pages9-10 Protein & Analisis ProteinM. Irfan FebriansyahNo ratings yet
- Oligonucleotide-Based Drugs and Therapeutics: Preclinical and Clinical Considerations for DevelopmentFrom EverandOligonucleotide-Based Drugs and Therapeutics: Preclinical and Clinical Considerations for DevelopmentNicolay FerrariNo ratings yet
- Ethylene Plant Contaminants: A Database and Interactive ToolDocument23 pagesEthylene Plant Contaminants: A Database and Interactive ToolEmmanuel LecrenayNo ratings yet
- Cells LifeDocument51 pagesCells LifeDileep KumarNo ratings yet
- Nitotile Epoxy GroutDocument2 pagesNitotile Epoxy GroutVincent JavateNo ratings yet
- Chemical Composition of The Mushroom Laetiporus Sulphureus (Bull.) MurillDocument8 pagesChemical Composition of The Mushroom Laetiporus Sulphureus (Bull.) MurillIvan R RamirezNo ratings yet
- Masterseal TC 254 - TdsDocument2 pagesMasterseal TC 254 - TdsHanyNo ratings yet
- 10 - Lipids: © 2013 W. H. Freeman and CompanyDocument71 pages10 - Lipids: © 2013 W. H. Freeman and Companymyatpwintp880No ratings yet
- Fluitest BIL Direct: Bilirubin Jendrassik/Gróf MethodDocument2 pagesFluitest BIL Direct: Bilirubin Jendrassik/Gróf MethodDarko MaksimovicNo ratings yet
- Some Basic Concepts of ChemistryDocument26 pagesSome Basic Concepts of ChemistryAnuj SharmaNo ratings yet
- STPSC20H12: 1200 V Power Schottky Silicon Carbide DiodeDocument8 pagesSTPSC20H12: 1200 V Power Schottky Silicon Carbide DiodeshivguptaNo ratings yet
- Formulary Sheet: Hair Conditioner, Premium, Split EndsDocument1 pageFormulary Sheet: Hair Conditioner, Premium, Split EndsShingi MameNo ratings yet
- Solving Word Problems in Engineering MathematicsDocument6 pagesSolving Word Problems in Engineering Mathematicsangelo diazNo ratings yet
- Remote Earth Cathodic ProtectionDocument2 pagesRemote Earth Cathodic ProtectionankiNo ratings yet
- Basic Design of Bna PlantDocument11 pagesBasic Design of Bna Plantmasprie2010100% (1)
- Book Chapter Microtomy Frozen Sections CryostatDocument15 pagesBook Chapter Microtomy Frozen Sections Cryostatbszool006No ratings yet
- Exp5 Baru..Document13 pagesExp5 Baru..ibrahimkhadijah100% (41)
- PPH Northshore Rolls Royce Specification Revision List 08-03-2021Document5 pagesPPH Northshore Rolls Royce Specification Revision List 08-03-2021MisterNoNo ratings yet
- Lewis Dot Structures and Molecular Geometries: Dr. WalkerDocument35 pagesLewis Dot Structures and Molecular Geometries: Dr. WalkerChristine Ano-os RoneNo ratings yet
- General Biology 1 (Light-Dependent Reactions)Document5 pagesGeneral Biology 1 (Light-Dependent Reactions)Jean DaclesNo ratings yet
- @StudyTime - Channel 11 - Sound (TH)Document12 pages@StudyTime - Channel 11 - Sound (TH)Legendary MathematicianNo ratings yet
- MMM7622S-Ficha TecnicaDocument14 pagesMMM7622S-Ficha TecnicaCarlos Alvarez BcNo ratings yet
- Gas Practice TestDocument10 pagesGas Practice TestTAHA GABRNo ratings yet
- Chemistry of Phosphorus in SoilDocument5 pagesChemistry of Phosphorus in SoilAbubakar Auwalu KwalamNo ratings yet
- 5 Ddcet Syllabus Engineering1702910382 - 231218 - 203535Document9 pages5 Ddcet Syllabus Engineering1702910382 - 231218 - 203535alonewarrior2004No ratings yet
- RAC (7th&8th) May2016 PDFDocument2 pagesRAC (7th&8th) May2016 PDFSachin MohalNo ratings yet
- Introduction To Inorganic ChemistryDocument11 pagesIntroduction To Inorganic ChemistryJianNo ratings yet
- Ecoquip 2 Eqm Vapor Abrasive Blast System: InstructionsDocument52 pagesEcoquip 2 Eqm Vapor Abrasive Blast System: InstructionsdimasjosesmithNo ratings yet
- Ingridient Function AIBDocument4 pagesIngridient Function AIBJaved Khan Nadir KhanNo ratings yet