You might also like
- DirectionalDocument113 pagesDirectionalNanthini Palanisamy100% (1)
- Komatsu PC130-7 Hydraulic Excavator Service ManualDocument342 pagesKomatsu PC130-7 Hydraulic Excavator Service ManualWilliam Bill Gabon100% (1)
- Yanmar Fault Codes ListDocument2 pagesYanmar Fault Codes ListJonas SvidinskasNo ratings yet
- TR50A ManualDocument372 pagesTR50A Manualrudyekajaya100% (1)
- X-Ray Diffraction Topography: International Series in the Science of the Solid StateFrom EverandX-Ray Diffraction Topography: International Series in the Science of the Solid StateNo ratings yet
- Guide For Suppliers Ext EnvDocument19 pagesGuide For Suppliers Ext EnvLuis APNo ratings yet
- Engineering Rock Mass Classification: Tunnelling, Foundations and LandslidesFrom EverandEngineering Rock Mass Classification: Tunnelling, Foundations and LandslidesRating: 4 out of 5 stars4/5 (5)
- Solar Electric Technician, Level-I SyllabusDocument28 pagesSolar Electric Technician, Level-I Syllabusrosan.sapkotaNo ratings yet
- Key GCC Projects To Be Awarded in The Next 6 MonthsDocument2 pagesKey GCC Projects To Be Awarded in The Next 6 MonthsDinesh Adwani100% (1)
- Approximation of Crystallite Size and Microstrain Via XRD Line Broadening Analysisin TiSiN ThinfilmsDocument6 pagesApproximation of Crystallite Size and Microstrain Via XRD Line Broadening Analysisin TiSiN ThinfilmsmustafaNo ratings yet
- Effect of The Nano Crystal Size On The X-Ray Diffraction Patterns of Biogenic Hydroxyapatite From Human, Bovine, and Porcine BonesDocument12 pagesEffect of The Nano Crystal Size On The X-Ray Diffraction Patterns of Biogenic Hydroxyapatite From Human, Bovine, and Porcine BonesAngga SaputraNo ratings yet
- 2D Mapping of Texture and Lattice Parameters of Dental EnamelDocument7 pages2D Mapping of Texture and Lattice Parameters of Dental EnamelHub SciNo ratings yet
- O Connell 2016Document5 pagesO Connell 2016Blessing HarvestNo ratings yet
- Imact of Defects On Optelectronic PropetiesDocument11 pagesImact of Defects On Optelectronic PropetiesSagar MoreNo ratings yet
- RV 5146Document16 pagesRV 5146Mr.WasgehtsiedasanNo ratings yet
- Powder DiffractionDocument9 pagesPowder DiffractionGary TrumpNo ratings yet
- Estimation of Lattice Strain in ZnO NanoparticlesDocument12 pagesEstimation of Lattice Strain in ZnO NanoparticleszuleNo ratings yet
- Ju et al 2015Document6 pagesJu et al 2015Marcijane VinhoteNo ratings yet
- Resume Dry Practical 1Document4 pagesResume Dry Practical 1Dharmapadmi KasilaniNo ratings yet
- PN 36 PDFDocument2 pagesPN 36 PDFPrasad RaikarNo ratings yet
- Heryanto_2018_J._Phys.__Conf._Ser._1080_012007Document6 pagesHeryanto_2018_J._Phys.__Conf._Ser._1080_012007hennaouisihamNo ratings yet
- Schofield 2002Document12 pagesSchofield 2002Wilhelm WesselsNo ratings yet
- Spectrochimica Acta Part A: Molecular and Biomolecular SpectrosDocument7 pagesSpectrochimica Acta Part A: Molecular and Biomolecular SpectrosBharath DNo ratings yet
- X-Ray Diffraction DissertationDocument7 pagesX-Ray Diffraction DissertationHelpOnWritingAPaperUK100% (1)
- Method For Determining Crystal Grain Size by X-RayDocument6 pagesMethod For Determining Crystal Grain Size by X-RayJulio César Huillca HuillcaNo ratings yet
- Artigo 7Document11 pagesArtigo 7Letícia SerrouNo ratings yet
- Dielectric Replica Measurement Revised Version CleanDocument22 pagesDielectric Replica Measurement Revised Version CleanMatthew DaviesNo ratings yet
- Nrod FlourenceDocument8 pagesNrod FlourenceSurinder SinghNo ratings yet
- RBD ConceptDocument14 pagesRBD ConceptAmar Bishram EkkaNo ratings yet
- The Details Related To A) Scientific MotivationDocument2 pagesThe Details Related To A) Scientific MotivationAmirtharajjmnNo ratings yet
- Dielectric Properties of Fe Doped Hydroxyapatite PreparedDocument8 pagesDielectric Properties of Fe Doped Hydroxyapatite PreparedVignesh RajaNo ratings yet
- Spectrochimica Acta Part A: Molecular and Biomolecular SpectrosDocument7 pagesSpectrochimica Acta Part A: Molecular and Biomolecular SpectrosJose Filipe Bacalhau RodriguesNo ratings yet
- Behavior of Mixture of Sulfamic Acid and Sodium Chloride: Dr. Rita A. Gharde, Divakar T. ChunarkarDocument4 pagesBehavior of Mixture of Sulfamic Acid and Sodium Chloride: Dr. Rita A. Gharde, Divakar T. ChunarkarIjarcet JournalNo ratings yet
- Spectroscopic Investigations of Carious Tooth DecayDocument6 pagesSpectroscopic Investigations of Carious Tooth DecaymustafaNo ratings yet
- 1 s2.0 S0254058419308181 MainDocument9 pages1 s2.0 S0254058419308181 MainRyka UsnilawatyNo ratings yet
- Band Gap Studies of Anatase Tio2 NanoparticlesDocument7 pagesBand Gap Studies of Anatase Tio2 Nanoparticlesnarayanan1701_318170No ratings yet
- Modified Scherrer EquationDocument8 pagesModified Scherrer EquationMiguel GuzmanNo ratings yet
- Tabela Que Explica Os Espectros Da Vermiculita No IRDocument9 pagesTabela Que Explica Os Espectros Da Vermiculita No IRDouglas SantosNo ratings yet
- Crystals 11 01050 v2Document7 pagesCrystals 11 01050 v2Giovanni Samir Perez HerreraNo ratings yet
- Thin Solid Films: S.R.C. Pinto, A.G. Rolo, A. Chahboun, R.J. Kashtiban, U. Bangert, M.J.M. GomesDocument4 pagesThin Solid Films: S.R.C. Pinto, A.G. Rolo, A. Chahboun, R.J. Kashtiban, U. Bangert, M.J.M. GomesNaseem chNo ratings yet
- Best practice in laser diffraction – a robustness studyDocument8 pagesBest practice in laser diffraction – a robustness studyfarageNo ratings yet
- Experimental Methods in Chemical Engineering: X-Ray Diffraction spectroscopy-XRDDocument13 pagesExperimental Methods in Chemical Engineering: X-Ray Diffraction spectroscopy-XRDNguyễn MaiNo ratings yet
- High Intensity X-Ray Diffraction in Transmission Mode Employing An Analog of Poisson's SpotDocument4 pagesHigh Intensity X-Ray Diffraction in Transmission Mode Employing An Analog of Poisson's Spotcristhian alvarezNo ratings yet
- Optical Scattering Properties of Phytoplankton: Measurements and Comparison of Various Species at Scattering Angles Between 1 U and 170uDocument6 pagesOptical Scattering Properties of Phytoplankton: Measurements and Comparison of Various Species at Scattering Angles Between 1 U and 170uGuanhua ZhouNo ratings yet
- Venu JCG2 2010Document6 pagesVenu JCG2 2010soma_venuNo ratings yet
- 2019 Canadian JoCE XRDDocument12 pages2019 Canadian JoCE XRDirem AydoganNo ratings yet
- Applied Radiation and Isotopes: Roseli K Unzel, Emico OkunoDocument4 pagesApplied Radiation and Isotopes: Roseli K Unzel, Emico OkunoEl Faqier IlallahNo ratings yet
- Optik 86Document6 pagesOptik 86z.umul9031No ratings yet
- 5 - Organic NLO Crystal - HMQTDocument8 pages5 - Organic NLO Crystal - HMQTSonal SaxenaNo ratings yet
- Normalizing XRF-scanner Data A Cautionary Note On The Interpretation of High-Resolution Records From Organic-Rich Lakes. Lowemark Et AlDocument7 pagesNormalizing XRF-scanner Data A Cautionary Note On The Interpretation of High-Resolution Records From Organic-Rich Lakes. Lowemark Et AlAlejandro Parra RojasNo ratings yet
- 1985 - Henderson Et Al. - Raman Spectra of Gallium and Germanium Substituted Silicate Glasses Variations in Intermediate Range Order.Document15 pages1985 - Henderson Et Al. - Raman Spectra of Gallium and Germanium Substituted Silicate Glasses Variations in Intermediate Range Order.Claudio BiaginiNo ratings yet
- First Laboratory X-Ray Diffraction Contrast Tomography For Grain Mapping of PolycrystalsDocument7 pagesFirst Laboratory X-Ray Diffraction Contrast Tomography For Grain Mapping of PolycrystalsXiaobing HuangNo ratings yet
- PHD Thesis On Raman SpectrosDocument6 pagesPHD Thesis On Raman Spectrosnadinebenavidezfortworth100% (2)
- Rietveld MethodDocument14 pagesRietveld MethodSek Wai KianNo ratings yet
- Synthesis and Characterisation of Hydroxyapatite Nanopowders by Hydrothermal MethodDocument7 pagesSynthesis and Characterisation of Hydroxyapatite Nanopowders by Hydrothermal Methodsalarphysics104No ratings yet
- XRD REVIEW PAPER Jeremiah MolaletsiDocument8 pagesXRD REVIEW PAPER Jeremiah MolaletsiJeremiah MolaletsiNo ratings yet
- Trager APB09 PDFDocument9 pagesTrager APB09 PDFmadrnessNo ratings yet
- R. Jerald Vijay Et Al. Journal of Crystal Growth 312 (2010) 420-425Document25 pagesR. Jerald Vijay Et Al. Journal of Crystal Growth 312 (2010) 420-425jerryremacNo ratings yet
- Journal of Physics and Chemistry of Solids: SciencedirectDocument5 pagesJournal of Physics and Chemistry of Solids: SciencedirectAnonymous dAN4dPGNo ratings yet
- Nlo Crystal Growth ThesisDocument7 pagesNlo Crystal Growth Thesislorieharrishuntsville100% (1)
- Amino-Grafted Water-Soluble Ferrimagnetic Iron Oxide Nanoparticles WithDocument9 pagesAmino-Grafted Water-Soluble Ferrimagnetic Iron Oxide Nanoparticles WithNadia Maulika Purnama WafiNo ratings yet
- Assessment of Growth of Silver Nanoparticles Synthesizedfrom An Ethylene Glycol-Silver Nitrate-Polyvinylpyrrolidone SolutionDocument11 pagesAssessment of Growth of Silver Nanoparticles Synthesizedfrom An Ethylene Glycol-Silver Nitrate-Polyvinylpyrrolidone SolutionMamata ManjariNo ratings yet
- Size Effects in The Raman Spectra of Tio Nanoparticles: Hyun Chul Choi, Young Mee Jung, Seung Bin KimDocument6 pagesSize Effects in The Raman Spectra of Tio Nanoparticles: Hyun Chul Choi, Young Mee Jung, Seung Bin Kimleoplasmo_201469720No ratings yet
- Oe 13 3 882Document7 pagesOe 13 3 882goyikon201No ratings yet
- XRD Analysis ThesisDocument6 pagesXRD Analysis ThesisBuyCheapPapersOnlineOmaha100% (2)
- Spectrochimica Acta Part A: Molecular and Biomolecular SpectrosDocument5 pagesSpectrochimica Acta Part A: Molecular and Biomolecular SpectrosPau VargasNo ratings yet
- Small Angle X-Ray and Neutron Scattering with Applications to GeomaterialsFrom EverandSmall Angle X-Ray and Neutron Scattering with Applications to GeomaterialsNo ratings yet
- 14 Purgadores Ayvaz PDFDocument13 pages14 Purgadores Ayvaz PDFjoaquin torrano veraNo ratings yet
- Zero Voltage Switch CA3059: ON SemiconductorDocument8 pagesZero Voltage Switch CA3059: ON SemiconductordangthanhlinhNo ratings yet
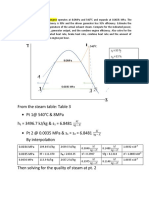
- From The Steam Table: Table 3 PT 1@ 540 C & 8mpa H 3496.7 KJ/KG & S 6.8481 PT 2 at 0.0035 Mpa & S S 6.8481 by InterpolationDocument5 pagesFrom The Steam Table: Table 3 PT 1@ 540 C & 8mpa H 3496.7 KJ/KG & S 6.8481 PT 2 at 0.0035 Mpa & S S 6.8481 by InterpolationMark AgusNo ratings yet
- Catalogo Soft StarterDocument16 pagesCatalogo Soft StarterEdwin Santiago Villegas AuquesNo ratings yet
- Question 1 MagnetismDocument44 pagesQuestion 1 MagnetismRanjit SinghNo ratings yet
- ForgingDocument56 pagesForgingZulfikarUdenNo ratings yet
- G-8 Choose The Letter of The Correct AnswerDocument3 pagesG-8 Choose The Letter of The Correct AnswerOrlando Hepulan BandolesNo ratings yet
- Sunlight Ozps Monoblock 2opzs100 12v 100a Ficha enDocument2 pagesSunlight Ozps Monoblock 2opzs100 12v 100a Ficha enRonald Victor Galarza HermitañoNo ratings yet
- NSF61Document12 pagesNSF61Congson Jeff100% (1)
- ATB Produktfolder EnglischDocument12 pagesATB Produktfolder EnglischonlineshaddowNo ratings yet
- Experience Up To 12 Days of Garden Freshness: Presenting Neo FreshDocument2 pagesExperience Up To 12 Days of Garden Freshness: Presenting Neo Freshrahul desaiNo ratings yet
- Yhf/Ge 12 Zxmaxaorx: Altair High Wall R-410A 60HzDocument4 pagesYhf/Ge 12 Zxmaxaorx: Altair High Wall R-410A 60HzBladimirNo ratings yet
- Lubrication Equipment Application Request: Fax Completed Form To 800-533-9656Document2 pagesLubrication Equipment Application Request: Fax Completed Form To 800-533-9656Instruktur MesinNo ratings yet
- 2.0 Psychrometry ApplicationDocument26 pages2.0 Psychrometry ApplicationMuhd ShazanyNo ratings yet
- Boomer S1 L-M PDFDocument4 pagesBoomer S1 L-M PDFArturo Pedro Salgado MedinaNo ratings yet
- Samsung RL42HDocument29 pagesSamsung RL42HMarcus MillerNo ratings yet
- Newton's Law of Motion and Forces: PHY01: General Physics 1 Senior High School DepartmentDocument26 pagesNewton's Law of Motion and Forces: PHY01: General Physics 1 Senior High School DepartmentcookingNo ratings yet
- ATYS dM3Document1 pageATYS dM3Reggie HarvirNo ratings yet
- Alp2 D22 EpDocument1 pageAlp2 D22 EpJefatura de Planta Invemet PeruNo ratings yet
- Design Considerations of Electrostatic Electrode e PDFDocument5 pagesDesign Considerations of Electrostatic Electrode e PDFTung ManhNo ratings yet
- Far Field Near Filed AntennaDocument3 pagesFar Field Near Filed Antennaanirban pahariNo ratings yet
- Extracting Water AirDocument9 pagesExtracting Water AirDeepak SharmaNo ratings yet
- Environmental Management in HotelsDocument6 pagesEnvironmental Management in Hotelsnguoibian99No ratings yet