You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Beta Anomaly An Ex-Ante Tail RiskDocument104 pagesBeta Anomaly An Ex-Ante Tail RiskDaniel Lee Eisenberg JacobsNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Why The Euro Will Rival The Dollar PDFDocument25 pagesWhy The Euro Will Rival The Dollar PDFDaniel Lee Eisenberg JacobsNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Claudia Jones Nuclear TestingDocument25 pagesClaudia Jones Nuclear TestingDaniel Lee Eisenberg JacobsNo ratings yet
- Continuity Change State of Process of Task ofDocument1 pageContinuity Change State of Process of Task ofDaniel Lee Eisenberg JacobsNo ratings yet
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
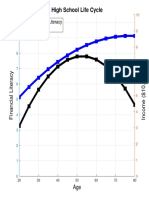
- High School Life Cycle: Financial Literacy IncomeDocument1 pageHigh School Life Cycle: Financial Literacy IncomeDaniel Lee Eisenberg JacobsNo ratings yet
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Theophilus Capital Against: Fisk Labor1Document9 pagesTheophilus Capital Against: Fisk Labor1Daniel Lee Eisenberg Jacobs100% (1)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Conference Group For Central European History of The American Historical AssociationDocument9 pagesConference Group For Central European History of The American Historical AssociationDaniel Lee Eisenberg JacobsNo ratings yet
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Individualization of Robo-AdviceDocument8 pagesIndividualization of Robo-AdviceDaniel Lee Eisenberg JacobsNo ratings yet
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Karl Kautsky Republic and Social Democra PDFDocument4 pagesKarl Kautsky Republic and Social Democra PDFDaniel Lee Eisenberg JacobsNo ratings yet
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Borel Sets PDFDocument181 pagesBorel Sets PDFDaniel Lee Eisenberg Jacobs100% (1)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Cover FERCDocument1 pageCover FERCDaniel Lee Eisenberg JacobsNo ratings yet
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- 1st International: Djacobs November 2020Document5 pages1st International: Djacobs November 2020Daniel Lee Eisenberg JacobsNo ratings yet
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Gpebook PDFDocument332 pagesGpebook PDFDaniel Lee Eisenberg JacobsNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Saving Behavior of Public Vocational High School Students of Business and Management Program in Semarang SitiDocument8 pagesThe Saving Behavior of Public Vocational High School Students of Business and Management Program in Semarang SitiDaniel Lee Eisenberg JacobsNo ratings yet
- Bank Loan Loss ProvisioningDocument17 pagesBank Loan Loss ProvisioningDaniel Lee Eisenberg JacobsNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Necessary and Sufficient Conditions For Dynamic OptimizationDocument18 pagesNecessary and Sufficient Conditions For Dynamic OptimizationDaniel Lee Eisenberg JacobsNo ratings yet
- Teach-In: Government of The People, by The People, For The PeopleDocument24 pagesTeach-In: Government of The People, by The People, For The PeopleDaniel Lee Eisenberg JacobsNo ratings yet
- Simple BeamerDocument25 pagesSimple BeamerDaniel Lee Eisenberg JacobsNo ratings yet
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- 5 6300732772877075609Document131 pages5 6300732772877075609Manoranjan sahuNo ratings yet
- T Cells - Production of T Cells - Types of T Cells - TeachMePhysiologyDocument3 pagesT Cells - Production of T Cells - Types of T Cells - TeachMePhysiologyNobodyNo ratings yet
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- CL1 HaemopoiesisDocument41 pagesCL1 HaemopoiesisSamuel George100% (1)
- The Ermgence of Meaning in Bio Systems - UriHershbergDocument89 pagesThe Ermgence of Meaning in Bio Systems - UriHershbergbroccoi66No ratings yet
- Autoimmune NeurologicalDocument385 pagesAutoimmune Neurologicalbodeadumitru9261No ratings yet
- Immunology Exam 4 Study GuideDocument2 pagesImmunology Exam 4 Study GuideKeaton EisenmengerNo ratings yet
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Lymphatic System Transes 1Document8 pagesLymphatic System Transes 1Sophia Margarette CawalingNo ratings yet
- Aid 2011 1502Document148 pagesAid 2011 1502pasargadae_mNo ratings yet
- Ijms 21 05223 PDFDocument42 pagesIjms 21 05223 PDFrohailNo ratings yet
- Cell Mediated and Humoral Immunity. Cytokines & The Immune Responses, Hypersensitivity ReactionsDocument40 pagesCell Mediated and Humoral Immunity. Cytokines & The Immune Responses, Hypersensitivity ReactionsAjaga RuqayyahNo ratings yet
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Quest II Answer Keys With ExplainationDocument23 pagesQuest II Answer Keys With Explainationbiotecnika_test75% (4)
- UT Dallas Syllabus For Biol4345.001 06s Taught by John Burr (Burr)Document1 pageUT Dallas Syllabus For Biol4345.001 06s Taught by John Burr (Burr)UT Dallas Provost's Technology GroupNo ratings yet
- Campbell Chapter 43 NotesDocument13 pagesCampbell Chapter 43 NotesRyan LiuNo ratings yet
- Exercise and The Regulation of Immune FunctionsDocument26 pagesExercise and The Regulation of Immune Functionskineucm2012No ratings yet
- M.Sc.-Microbiology Syllabus HNBDocument58 pagesM.Sc.-Microbiology Syllabus HNBBiology Made EasyNo ratings yet
- Frog Immune System Robert2016Document7 pagesFrog Immune System Robert2016andreNo ratings yet
- ASmaDocument48 pagesASmaKelvin LaibidaNo ratings yet
- Human Health and DiseaseDocument162 pagesHuman Health and DiseaseKanmani SathiachandranNo ratings yet
- 2014 - MesenchymalDocument18 pages2014 - MesenchymalMiguel ÁngelNo ratings yet
- Fundamentals of Immunology (Updated)Document59 pagesFundamentals of Immunology (Updated)Marydith OrtilloNo ratings yet
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- 11.2 Development of ImmunityDocument90 pages11.2 Development of ImmunityDaksha yashaNo ratings yet
- Abraham and Medzhitov 2011 - Interactions Between The Host Innate Immune System and Microbes in Inflammatory Bowel DiseaseDocument9 pagesAbraham and Medzhitov 2011 - Interactions Between The Host Innate Immune System and Microbes in Inflammatory Bowel DiseaseAchille BroggiNo ratings yet
- Introduction To The Immune System: Department of Biotechnology Ramaiah University of Applied Sciences BangaloreDocument86 pagesIntroduction To The Immune System: Department of Biotechnology Ramaiah University of Applied Sciences Bangaloreprathyoosha baskaran100% (1)
- JC-2 Tutorial-1 Immunology Practice MCQ'sDocument14 pagesJC-2 Tutorial-1 Immunology Practice MCQ'shelamahjoubmounirdmo100% (5)
- Immune Response To TumorDocument14 pagesImmune Response To TumorFebrian Alfaro100% (1)
- RadVacWhite Paper SARS CoV 2 Vaccine Ver 3 3 1Document69 pagesRadVacWhite Paper SARS CoV 2 Vaccine Ver 3 3 1Karoline MarxNo ratings yet
- Kuby Immunology TransplantationDocument21 pagesKuby Immunology TransplantationMalki kawtarNo ratings yet
- Immuno TherapyDocument22 pagesImmuno TherapyLiliana GuzmánNo ratings yet
- HKDSE Biology Part 3 Health & DiseasesDocument17 pagesHKDSE Biology Part 3 Health & DiseasesTSZ YAN CHEUNGNo ratings yet
- ImmunologyDocument92 pagesImmunology[161]Shuaib AktherNo ratings yet
- 10% Human: How Your Body's Microbes Hold the Key to Health and HappinessFrom Everand10% Human: How Your Body's Microbes Hold the Key to Health and HappinessRating: 4 out of 5 stars4/5 (33)
- The Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceFrom EverandThe Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceRating: 4.5 out of 5 stars4.5/5 (517)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (5)