You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (894)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (587)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Work Item Description UOM Scope Achieved Progress Planned Start Actual Start Planned Finish Actual Finish RemarksDocument2 pagesWork Item Description UOM Scope Achieved Progress Planned Start Actual Start Planned Finish Actual Finish Remarksnishant361No ratings yet
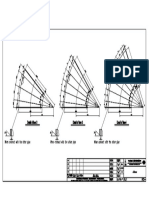
- Tank JackingDocument1 pageTank Jackingnishant361No ratings yet
- Coal Mill Field Quality PlanDocument3 pagesCoal Mill Field Quality Plannishant361No ratings yet
- Contractor Safety RequirementsDocument32 pagesContractor Safety Requirementsask1400No ratings yet
- Chapter 5CDocument6 pagesChapter 5Cnishant361No ratings yet
- Hydrogen Cracks in Steels TWIDocument5 pagesHydrogen Cracks in Steels TWInishant361No ratings yet
- Elbow For Dedusting DuctDocument1 pageElbow For Dedusting Ductnishant361No ratings yet
- Structural Steel ErectionDocument17 pagesStructural Steel ErectionHemanth KumarNo ratings yet
- Flange TypesDocument1 pageFlange TypesHadi UtomoNo ratings yet
- Dimension Tolerance (Coal Mill)Document4 pagesDimension Tolerance (Coal Mill)nishant361No ratings yet
- ID FanDocument4 pagesID Fannishant361No ratings yet
- Pressure Testing SafetyDocument4 pagesPressure Testing Safetyapi-26143956100% (2)
- Chapter 1Document6 pagesChapter 1D.RameshkumarNo ratings yet
- CoatingDocument8 pagesCoatingAbhinav GoyalNo ratings yet
- English - Compact Inspector's ManualDocument16 pagesEnglish - Compact Inspector's Manualnishant361No ratings yet
- En 12068Document48 pagesEn 12068pametnjakovicmladji100% (12)
- Bhel Welding GuidelinesDocument64 pagesBhel Welding Guidelinesashutoshsachan100% (3)
- VTG Iom 2013 0815Document96 pagesVTG Iom 2013 0815nishant361No ratings yet
- 10 - RT SG IiDocument120 pages10 - RT SG Iinishant361No ratings yet
- Chapter 10 Radiography in Modern IndustryDocument17 pagesChapter 10 Radiography in Modern Industrynishant361No ratings yet
- Mess Bill For The Month of SEPTEMBER-2012: TotalDocument2 pagesMess Bill For The Month of SEPTEMBER-2012: Totalnishant361No ratings yet
- Heat Treatment ManualDocument25 pagesHeat Treatment Manualraj101086100% (1)
- Chapter 11 Radiography in Modern IndustryDocument7 pagesChapter 11 Radiography in Modern Industrynishant361No ratings yet
- Real Time Radiography Course BookletDocument48 pagesReal Time Radiography Course BookletAnonymous gFcnQ4goNo ratings yet
- Pure Protect BrochureDocument6 pagesPure Protect Brochuredhuaraaj212No ratings yet
- Chapter 4 Radiography in Modern IndustryDocument11 pagesChapter 4 Radiography in Modern Industrynishant361No ratings yet
- Heat Treatment ManualDocument25 pagesHeat Treatment Manualraj101086100% (1)
- 1booked Tickets History PDFDocument1 page1booked Tickets History PDFnishant361No ratings yet
- IndiaMike PDFDocument7 pagesIndiaMike PDFnishant361No ratings yet
- Hydrogen Energy, Economy and StorageDocument15 pagesHydrogen Energy, Economy and StorageLe NhanNo ratings yet
- Hidrogen Plasma Reactors 1-s2.0-S036031992204839X-mainDocument16 pagesHidrogen Plasma Reactors 1-s2.0-S036031992204839X-mainsolutronicNo ratings yet
- Dessertation FINALDocument16 pagesDessertation FINALeshaan guptaNo ratings yet
- Master On Hydrogen ProcessesDocument199 pagesMaster On Hydrogen ProcessesAhmed MohamedNo ratings yet
- Petersen mst15Document8 pagesPetersen mst15api-271093465No ratings yet
- Recent Advances in Liquid Organic Hydrogen Carriers: An Alcohol-Based Hydrogen EconomyDocument15 pagesRecent Advances in Liquid Organic Hydrogen Carriers: An Alcohol-Based Hydrogen EconomySajid Mohy Ul Din100% (1)
- Hydrogen OSTIDocument40 pagesHydrogen OSTIAnurag BholeNo ratings yet
- 4 A+Green+Spider+projectDocument15 pages4 A+Green+Spider+projectJuan Carlos Villar RodriguezNo ratings yet
- Progress Report For Hydrogen, Fuel Cells, and Infrastructure Technologies Program (DOE, 2020) (T) PDFDocument649 pagesProgress Report For Hydrogen, Fuel Cells, and Infrastructure Technologies Program (DOE, 2020) (T) PDFShiva KumarNo ratings yet
- Abstract Book: INSEF Regional Fair - ChennaiDocument28 pagesAbstract Book: INSEF Regional Fair - ChennaiKathir VelNo ratings yet
- Hydrogenation Properties of Mg-Al AlloysDocument25 pagesHydrogenation Properties of Mg-Al Alloysandr1976No ratings yet
- CompositeMaterials PHD Summary-2007Document31 pagesCompositeMaterials PHD Summary-2007Ege menNo ratings yet
- Semester IDocument17 pagesSemester IMohit PadheeNo ratings yet
- Muhammad Rashid Usman, PHDDocument9 pagesMuhammad Rashid Usman, PHDTanveerNo ratings yet
- Co-Ni Supported Yttrium Oxide Material As A Catalyst For Ammonia Decomposition To CO - Free HydrogenDocument12 pagesCo-Ni Supported Yttrium Oxide Material As A Catalyst For Ammonia Decomposition To CO - Free HydrogenLekidelu AsratNo ratings yet
- Robert A. Varin, Tom Czujko, Eric B. Wasmund, Zbigniew S. WronskiDocument15 pagesRobert A. Varin, Tom Czujko, Eric B. Wasmund, Zbigniew S. WronskiMartin Fibela EsparzaNo ratings yet
- Current Technologies and Challenges of Applying Fuel Cell Hybrid PropulsionDocument22 pagesCurrent Technologies and Challenges of Applying Fuel Cell Hybrid PropulsionTomislav RosuljNo ratings yet
- Development of Standards For Hydrogen SafetyDocument4 pagesDevelopment of Standards For Hydrogen SafetyJesusNo ratings yet
- Usman-2013-Dehydrogenation of MethylcyclohexaneDocument8 pagesUsman-2013-Dehydrogenation of MethylcyclohexaneRashidNo ratings yet
- J Ijhydene 2013 07 016Document8 pagesJ Ijhydene 2013 07 016Dharun TNo ratings yet
- Development of High Pressure Hydrogen Storage Tank For Storage and Gaseous Truck DeliveryDocument26 pagesDevelopment of High Pressure Hydrogen Storage Tank For Storage and Gaseous Truck DeliveryFederico CasconNo ratings yet
- Advanced Materials For Hydrogen ProductionDocument150 pagesAdvanced Materials For Hydrogen ProductionGhazanfar SheikhNo ratings yet
- Hydrogen StorageDocument9 pagesHydrogen StorageLisa DiasNo ratings yet
- Hydrogen Storage - Department of EnergyDocument7 pagesHydrogen Storage - Department of EnergyJulian Laureano LopezNo ratings yet
- IHTEC 2019 Program Booklet v13Document14 pagesIHTEC 2019 Program Booklet v13fficici0% (1)
- Review Zuettel 2004Document17 pagesReview Zuettel 2004Saki ShaikhNo ratings yet
- First-Principles Study On The Dehydrogenation Thermodynamics and Kinetics of Ti, ZR, V and NB Doped MgH2Document10 pagesFirst-Principles Study On The Dehydrogenation Thermodynamics and Kinetics of Ti, ZR, V and NB Doped MgH2kienfinanceNo ratings yet
- Nygård 2019Document9 pagesNygård 2019Wágner B SilvaNo ratings yet
- AFP TankDocument17 pagesAFP TankAlexandra PetreNo ratings yet
- 982359Document352 pages982359Sir TemplarNo ratings yet