You might also like
- 68th AACC Annual Scientific Meeting Abstract eBookFrom Everand68th AACC Annual Scientific Meeting Abstract eBookNo ratings yet
- Marinesco-Sjo Gren Syndrome - 2013Document5 pagesMarinesco-Sjo Gren Syndrome - 2013Irfan RazaNo ratings yet
- Epilepsia EpicongressDocument245 pagesEpilepsia EpicongressВасилий КоптеловNo ratings yet
- Morphometry of WS On ASDocument6 pagesMorphometry of WS On ASAdrien DuthéNo ratings yet
- Novel p53 Splicing Site Mutation in Li-Fraumeni-like Syndrome With OsteosarcomaDocument5 pagesNovel p53 Splicing Site Mutation in Li-Fraumeni-like Syndrome With OsteosarcomaMissy MishaNo ratings yet
- (1479683X - European Journal of Endocrinology) Parental Genomic Imprinting in EndocrinopathiesDocument9 pages(1479683X - European Journal of Endocrinology) Parental Genomic Imprinting in EndocrinopathiesRavia SharmaNo ratings yet
- 23 - 224Praktis-Strategi Terapi Cairan Pada DehidrasiDocument8 pages23 - 224Praktis-Strategi Terapi Cairan Pada DehidrasiRahmat AbbasNo ratings yet
- Batten Disease: Features To Facilitate Early Diagnosis: Scientific ReportDocument6 pagesBatten Disease: Features To Facilitate Early Diagnosis: Scientific ReportMaferNo ratings yet
- Biopolym - Cell 2018 34 5 361 enDocument6 pagesBiopolym - Cell 2018 34 5 361 enАнна ШаповаловаNo ratings yet
- Biopolym - Cell 2018 34 5 361 enDocument6 pagesBiopolym - Cell 2018 34 5 361 enАнна ШаповаловаNo ratings yet
- Methylation, Cytogenetic and Fish Tests in The Molecular Diagnosis of Prader-Willi and Angelman SyndromesDocument12 pagesMethylation, Cytogenetic and Fish Tests in The Molecular Diagnosis of Prader-Willi and Angelman SyndromesNayara MacedoNo ratings yet
- 10 1038@nature07261Document7 pages10 1038@nature07261ANo ratings yet
- Fragile X Syndrome Due to a Rare Missense MutationDocument5 pagesFragile X Syndrome Due to a Rare Missense MutationhanzelNo ratings yet
- Fragile X Syndrome (Martin-Bell Syndrome)Document6 pagesFragile X Syndrome (Martin-Bell Syndrome)yudhi kurniawanNo ratings yet
- Gim 200269 ADocument5 pagesGim 200269 AfakeempireNo ratings yet
- Genodermatosis MCQsDocument152 pagesGenodermatosis MCQsDr.Tawheed88% (8)
- AngelmanDocument12 pagesAngelmanapi-295680726No ratings yet
- Full TextDocument4 pagesFull TextladybieibiNo ratings yet
- (03241750 - Acta Medica Bulgarica) Molecular-Genetic Diagnostics of Angelman Syndrome - The Bulgarian ExperienceDocument8 pages(03241750 - Acta Medica Bulgarica) Molecular-Genetic Diagnostics of Angelman Syndrome - The Bulgarian ExperienceTeodorNo ratings yet
- Recurrent 7q11.23 Deletions Identified in Patients with Neurodevelopmental DisordersDocument9 pagesRecurrent 7q11.23 Deletions Identified in Patients with Neurodevelopmental DisordersEAPNo ratings yet
- Clinical Features and Gene Mutational Spectrum of CDKL5-related Diseases in A Cohort of Chinese PatientsDocument7 pagesClinical Features and Gene Mutational Spectrum of CDKL5-related Diseases in A Cohort of Chinese PatientsGaly Crt FlzNo ratings yet
- A TESIS GalvanDocument28 pagesA TESIS GalvanSaul PsiqNo ratings yet
- 439 2007 Article 362Document6 pages439 2007 Article 362Zinik EşanuNo ratings yet
- MLPA Dummy ReportDocument3 pagesMLPA Dummy ReportAakash verma100% (1)
- 2.3kb FOXG1 gene deletion detected by array CGHDocument1 page2.3kb FOXG1 gene deletion detected by array CGHdvNo ratings yet
- Journal of Child Neurology 2Document7 pagesJournal of Child Neurology 2NEUROLAB ESCALÓNNo ratings yet
- Patogenesis IMNDocument23 pagesPatogenesis IMNshiloinNo ratings yet
- Menkes Kinky Hair Disease (Menkes Syndrome) - A Case ReportDocument5 pagesMenkes Kinky Hair Disease (Menkes Syndrome) - A Case ReportTannov SiregarNo ratings yet
- ng00229 PDFDocument3 pagesng00229 PDFPaijo SusenoNo ratings yet
- Role For Hedgehog Signaling in Cranial-Suture Development and ObesityDocument9 pagesRole For Hedgehog Signaling in Cranial-Suture Development and ObesityDanny Alvarez FocacciNo ratings yet
- Prion (CJD), Lahiru Perera, 23416785Document6 pagesPrion (CJD), Lahiru Perera, 23416785Lahiru PereraNo ratings yet
- McKinnis Bone Marrow Transplantation Hunter Syndrome The Journal of PediatricsDocument17 pagesMcKinnis Bone Marrow Transplantation Hunter Syndrome The Journal of PediatricsBoNo ratings yet
- 2008 Elsea EurJHumGenetDocument10 pages2008 Elsea EurJHumGenetEAPNo ratings yet
- NPM1 Gene Deletions in MDS Patients with 5q- Deletion and Complex KaryotypeDocument2 pagesNPM1 Gene Deletions in MDS Patients with 5q- Deletion and Complex KaryotypeglodovichiNo ratings yet
- 61-1-13sd RettDocument11 pages61-1-13sd RettCarolina GuzmánNo ratings yet
- A Novel RLIM/RNF12 Variant Disrupts Protein Stability and Function To Cause Severe Tonne-Kalscheuer SyndromeDocument9 pagesA Novel RLIM/RNF12 Variant Disrupts Protein Stability and Function To Cause Severe Tonne-Kalscheuer SyndromeMohamad SoveyziNo ratings yet
- De Novo Mutations in YWHAG Cause Early Onset EpilepsyDocument11 pagesDe Novo Mutations in YWHAG Cause Early Onset EpilepsyGiselle Costa Daniel HonoratoNo ratings yet
- EJE-Cuny2013Document9 pagesEJE-Cuny2013W Antonio Muñoz ChNo ratings yet
- Nascimento Et Al. - 2012 - Adrenoleukodystrophy A Forgotten Diagnosis in Children With Primary Addison' S DiseaseDocument5 pagesNascimento Et Al. - 2012 - Adrenoleukodystrophy A Forgotten Diagnosis in Children With Primary Addison' S DiseaseflashjetNo ratings yet
- CHAP NO4 Results (Abbas) Rewritten FileDocument12 pagesCHAP NO4 Results (Abbas) Rewritten FileRahid KhanNo ratings yet
- Spinal Muscular Atrophy 2023Document21 pagesSpinal Muscular Atrophy 2023Arbey Aponte PuertoNo ratings yet
- Mutations in Mitochondr Ial GenesDocument6 pagesMutations in Mitochondr Ial Geneslynden matbaganNo ratings yet
- FRAGILE X SYNDROME: CLINICAL FEATURES AND MOLECULAR BASISDocument65 pagesFRAGILE X SYNDROME: CLINICAL FEATURES AND MOLECULAR BASISFlory ZapantaNo ratings yet
- La Epigenética y Los Estudios en Gemelos en El Campo de La PsiquiatríaDocument9 pagesLa Epigenética y Los Estudios en Gemelos en El Campo de La PsiquiatríaHerb MedrNo ratings yet
- Pyruvate Kinase Deficiency and Malaria: Brief ReportDocument6 pagesPyruvate Kinase Deficiency and Malaria: Brief ReportErlangga Perwira NegaraNo ratings yet
- AP1S2 Is Mutated in Xlinked DandyWalker Malformation With Intellectual Disability Basal Ganglia Disease and Seizures Pettigrew Syndrome - 2014Document6 pagesAP1S2 Is Mutated in Xlinked DandyWalker Malformation With Intellectual Disability Basal Ganglia Disease and Seizures Pettigrew Syndrome - 2014Jose Rafael Villafan BernalNo ratings yet
- Book JewsDocument230 pagesBook JewsFred Duarte CaldeiraNo ratings yet
- Reyes - Unit 2 (Part 2)Document4 pagesReyes - Unit 2 (Part 2)Justine Ericca ReyesNo ratings yet
- Genotype-Phenotype Studies in Three Families PQBP1Document9 pagesGenotype-Phenotype Studies in Three Families PQBP1Mosabbira RahmanNo ratings yet
- Genetic Mutations BTCIDocument59 pagesGenetic Mutations BTCIDharti Adhia100% (1)
- Ocular Features of Marfan Syndrome: Gordana Stanković-Babić, Milena Vujanović, Jasmina Đorđević-Jocić, Sonja CekićDocument4 pagesOcular Features of Marfan Syndrome: Gordana Stanković-Babić, Milena Vujanović, Jasmina Đorđević-Jocić, Sonja CekićmongiiiNo ratings yet
- Progressive Increase of The Mutated Mitochondria1 DNA Fraction in Kearns-Sayre SyndromeDocument6 pagesProgressive Increase of The Mutated Mitochondria1 DNA Fraction in Kearns-Sayre SyndromeGréta BotyánszkiNo ratings yet
- 05 Molecular Genetics of The EpilepsiesDocument15 pages05 Molecular Genetics of The EpilepsiesvenkatesannagarajanNo ratings yet
- Microscopy Res Technique - 2001 - Southwood - Molecular pathways of oligodendrocyte apoptosis revealed by mutations inDocument9 pagesMicroscopy Res Technique - 2001 - Southwood - Molecular pathways of oligodendrocyte apoptosis revealed by mutations inAnglia LopesNo ratings yet
- Detect 80% of AS Cases with Methylation AnalysisDocument5 pagesDetect 80% of AS Cases with Methylation AnalysisBeatriz AzevedoNo ratings yet
- 2008 - MarshallDocument12 pages2008 - MarshallCarlosNo ratings yet
- Genetic and Neurodevelopmental Spectrum of SYNGAP1-associated Intellectual Disability and EpilepsyDocument12 pagesGenetic and Neurodevelopmental Spectrum of SYNGAP1-associated Intellectual Disability and EpilepsySaya OtonashiNo ratings yet
- Winawer Et Al-2018-Annals of NeurologyDocument14 pagesWinawer Et Al-2018-Annals of NeurologyAndoingNo ratings yet
- Neuroendocrine Tumors: Surgical Evaluation and ManagementFrom EverandNeuroendocrine Tumors: Surgical Evaluation and ManagementJordan M. CloydNo ratings yet
- ContentServer AspDocument12 pagesContentServer AspAkmal NugrahaNo ratings yet
- ContentServer AspDocument12 pagesContentServer AspAkmal NugrahaNo ratings yet
- Idr 7 323Document7 pagesIdr 7 323Akmal NugrahaNo ratings yet
- Idr 7 323Document7 pagesIdr 7 323Akmal NugrahaNo ratings yet
- ContentServer AspDocument12 pagesContentServer AspAkmal NugrahaNo ratings yet
- Idr 7 323Document7 pagesIdr 7 323Akmal NugrahaNo ratings yet
- RefraksiDocument53 pagesRefraksigilaliNo ratings yet
- RefraksiDocument53 pagesRefraksigilaliNo ratings yet
- CERAMAHDocument2 pagesCERAMAHAkmal NugrahaNo ratings yet
- 02 - Physical ExaminationDocument69 pages02 - Physical Examinationkrizia_arifinNo ratings yet
- CERAMAHDocument2 pagesCERAMAHAkmal NugrahaNo ratings yet
- CERAMAHDocument2 pagesCERAMAHAkmal NugrahaNo ratings yet
- Chromatin RemodellingDocument234 pagesChromatin Remodellingplastioid4079No ratings yet
- Aberrant Astrocyte Protein Secretion Contributes To Altered Neuronal Development in Multiple Models of Neurodevelopmental DisordersDocument36 pagesAberrant Astrocyte Protein Secretion Contributes To Altered Neuronal Development in Multiple Models of Neurodevelopmental DisordersLeon PalomeraNo ratings yet
- Combinepdf 7Document129 pagesCombinepdf 7Joe JosephNo ratings yet
- HMB200 Lecture 10 ASD Genetics 2020-21Document56 pagesHMB200 Lecture 10 ASD Genetics 2020-21bluetooth opencvNo ratings yet
- Evaluation of the Child with Global Developmental Delay: Diagnostic Yield of Metabolic and Genetic InvestigationsDocument61 pagesEvaluation of the Child with Global Developmental Delay: Diagnostic Yield of Metabolic and Genetic InvestigationsAnsilNo ratings yet
- FDAapproves Daybuethefirsttreatmentfor RettsyndromeDocument8 pagesFDAapproves Daybuethefirsttreatmentfor RettsyndromeNeethu Anna StephenNo ratings yet
- Rett Syndrome A Neurological Disorder With MetabolDocument17 pagesRett Syndrome A Neurological Disorder With MetabolTiara PuspaNo ratings yet
- Avexis Investor PresentationDocument20 pagesAvexis Investor PresentationmedtechyNo ratings yet
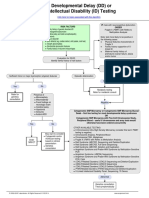
- Developmental Delay (DD) or Intellectual Disability (ID) Testing AlgorithmDocument1 pageDevelopmental Delay (DD) or Intellectual Disability (ID) Testing AlgorithmNeo Mervyn Monaheng100% (1)
- Autism Spectrum DisordersDocument9 pagesAutism Spectrum DisordersClaudia Verónica Revilla PazNo ratings yet
- Prenatal ScreningDocument44 pagesPrenatal ScreningHoopmen tampubolonNo ratings yet
- Science:, 792 (2003) Robin D. Rogers and Kenneth R. SeddonDocument3 pagesScience:, 792 (2003) Robin D. Rogers and Kenneth R. SeddonJohnSmithNo ratings yet
- Rett SyndromeDocument13 pagesRett SyndromeAileish Kate Lanuevo JaudianNo ratings yet
- Thesis TitlesDocument25 pagesThesis TitlesMicah Dianne DizonNo ratings yet
- Rett Syndrome 1Document19 pagesRett Syndrome 1api-545157726No ratings yet
- PHD Credit Seminar 2 Punjab Agricultural University, LudhianaDocument141 pagesPHD Credit Seminar 2 Punjab Agricultural University, LudhianaSUSHANT DHARNo ratings yet
- What Is Rett Syndrome?Document4 pagesWhat Is Rett Syndrome?Jon_NamikazeNo ratings yet
- Rett SyndromeDocument55 pagesRett SyndromeSnezana MihajlovicNo ratings yet
- Intellectual Disability in Children - Evaluation For A Cause - UpToDateDocument30 pagesIntellectual Disability in Children - Evaluation For A Cause - UpToDatecapt_zoe100% (1)
- THE Ironies of Human Mind: A Case of Rett SyndromeDocument5 pagesTHE Ironies of Human Mind: A Case of Rett Syndromeaprillia kusuma putriNo ratings yet
- Athens 2010 IALP - PROCEEDINGS PDFDocument882 pagesAthens 2010 IALP - PROCEEDINGS PDFYelly Andriani Barlian100% (1)
- Neural and Synaptic Defects in Autism SPDocument287 pagesNeural and Synaptic Defects in Autism SPGabriel NeiraNo ratings yet
- The Role of Neuroglia in AutismDocument30 pagesThe Role of Neuroglia in AutismWesley M.SantosNo ratings yet
- Autism A Neuroepigenetic DisorderDocument13 pagesAutism A Neuroepigenetic DisorderMelania TonelloNo ratings yet
- Antipurinergic Therapy For Autism-An In-Depth Review.Document15 pagesAntipurinergic Therapy For Autism-An In-Depth Review.Miguel Romero100% (1)
- Abstracts From ESMED Congress 2021Document197 pagesAbstracts From ESMED Congress 2021European Society of Medicine (ESMED)100% (2)
- Pediatric Neurology - Lawson Peter N. (SRG)Document200 pagesPediatric Neurology - Lawson Peter N. (SRG)Hriday DeNo ratings yet
- Genetic Causes of Syndromic and Non-Syndromic Autism PDFDocument9 pagesGenetic Causes of Syndromic and Non-Syndromic Autism PDFRaisa CoppolaNo ratings yet
- Retraso Global Del DesarrolloDocument18 pagesRetraso Global Del Desarrolloenypaola19No ratings yet
- GABAA receptor downregulation in autism brain regionsDocument7 pagesGABAA receptor downregulation in autism brain regionsaini qurrotullainNo ratings yet