You might also like
- How To Outline A Novel Step by StepDocument7 pagesHow To Outline A Novel Step by StepgunjanNo ratings yet
- The One Page Novel Scene SpreadsheetDocument5 pagesThe One Page Novel Scene SpreadsheetLittle_Wing67No ratings yet
- Prospectus 2020-21Document138 pagesProspectus 2020-21gunjanNo ratings yet
- Administrative Institutions in India: Biyani's Think TankDocument69 pagesAdministrative Institutions in India: Biyani's Think TankgunjanNo ratings yet
- List of The Most Popular Given Names in South Korea - Wikipedia PDFDocument25 pagesList of The Most Popular Given Names in South Korea - Wikipedia PDFgunjanNo ratings yet
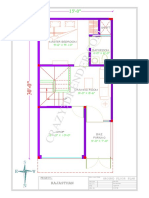
- Ground Floor Plan 22 PDFDocument1 pageGround Floor Plan 22 PDFgunjanNo ratings yet
- ThePoemsofSirWalterRaleigh 10154352 PDFDocument294 pagesThePoemsofSirWalterRaleigh 10154352 PDFgunjanNo ratings yet
- Ground Floor Plan 22 PDFDocument1 pageGround Floor Plan 22 PDFgunjanNo ratings yet
- First Floor Plan 15 PDFDocument1 pageFirst Floor Plan 15 PDFgunjanNo ratings yet
- FDocument19 pagesFTulis Tyoriny IrwantoNo ratings yet
- The Grandmaster of Demonic Cultivation Book One Mo Xiang Tong XiaDocument430 pagesThe Grandmaster of Demonic Cultivation Book One Mo Xiang Tong XiaShin Keichi100% (6)
- First Floor Plan 15 PDFDocument1 pageFirst Floor Plan 15 PDFgunjanNo ratings yet
- Lou Gehrig S Disease en inDocument2 pagesLou Gehrig S Disease en ingunjanNo ratings yet
- Ground Floor Plan 22 PDFDocument1 pageGround Floor Plan 22 PDFgunjanNo ratings yet
- Lelouch Lamperouge PDFDocument8 pagesLelouch Lamperouge PDFgunjan100% (1)
- B.sc. (Hons.) Nursing Acknowledgement CardDocument1 pageB.sc. (Hons.) Nursing Acknowledgement CardgunjanNo ratings yet
- The Grandmaster of Demonic Cultivation Book One Mo Xiang Tong XiaDocument430 pagesThe Grandmaster of Demonic Cultivation Book One Mo Xiang Tong XiaShin Keichi100% (6)
- Curriculam Vitae: Ajit Kumar PathakDocument2 pagesCurriculam Vitae: Ajit Kumar PathakgunjanNo ratings yet
- Curriculum Vitae: Career ObjectiveDocument2 pagesCurriculum Vitae: Career ObjectivegunjanNo ratings yet
- Presentation 1Document1 pagePresentation 1gunjanNo ratings yet
- Prestige ScriptDocument128 pagesPrestige ScriptMichael Kosciesza100% (1)
- My Name Is Daisy NagarkotiDocument1 pageMy Name Is Daisy NagarkotigunjanNo ratings yet
- Curriculam Vitae: Ajit Kumar PathakDocument2 pagesCurriculam Vitae: Ajit Kumar PathakgunjanNo ratings yet
- Circulatory Motion ExplainedDocument2 pagesCirculatory Motion ExplainedgunjanNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (73)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (399)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (344)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (119)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- DOH Updated Programs A-FDocument61 pagesDOH Updated Programs A-Fdeeday echavezNo ratings yet
- Bio Attack Alert: The Strecker MemorandumDocument12 pagesBio Attack Alert: The Strecker MemorandumCazzac111100% (1)
- Disease and Astrology PDFDocument9 pagesDisease and Astrology PDFAbhishek P BenjaminNo ratings yet
- Volume 46, Issue 6, February 6, 2015Document44 pagesVolume 46, Issue 6, February 6, 2015BladeNo ratings yet
- C11 P07 National Aids Control ProgrammeDocument32 pagesC11 P07 National Aids Control ProgrammeChandana KrishnaNo ratings yet
- Test Bank For Social Problems 14e 14th Edition William Kornblum Joseph JulianDocument17 pagesTest Bank For Social Problems 14e 14th Edition William Kornblum Joseph JulianMohammad Brazier100% (18)
- Clinical Manifestation of Oral TuberculosisDocument6 pagesClinical Manifestation of Oral TuberculosisSasa AprilaNo ratings yet
- BSMC-Science Catalogue PDFDocument48 pagesBSMC-Science Catalogue PDFJatin Pahuja0% (1)
- Certified Coding Specialist CCS Exam Preparation OCRDocument319 pagesCertified Coding Specialist CCS Exam Preparation OCRKian Gonzaga100% (32)
- Health9 - q2 - Mod1 - Drug Scenario in The PhilippinesDocument32 pagesHealth9 - q2 - Mod1 - Drug Scenario in The PhilippinesMaria Darrilyn Capinig100% (7)
- Assessment of Knowledge, Attitude Andpractice Toward Sexually Transmitteddiseases in Boditi High School StudentsDocument56 pagesAssessment of Knowledge, Attitude Andpractice Toward Sexually Transmitteddiseases in Boditi High School StudentsMinlik-alew Dejenie88% (8)
- Case 37-2020: A 35-Year-Old Man With Lymphadenopathy and PetechiaeDocument11 pagesCase 37-2020: A 35-Year-Old Man With Lymphadenopathy and PetechiaePatriciaNo ratings yet
- HIV and AIDS - Basic Facts: GeneralDocument18 pagesHIV and AIDS - Basic Facts: GeneralBudoy WashupapiNo ratings yet
- Imci Chart Booklet 2011 - 1 PDFDocument64 pagesImci Chart Booklet 2011 - 1 PDFDanny Irung100% (2)
- Application Form 2019 PepfarDocument6 pagesApplication Form 2019 PepfarCollins OsanyoNo ratings yet
- U01 Apps PHSunSMART PHSunSMARTFiles SymbiosysFiles Generated OutputSIPDF 098692O090520182219005228644 1525875549108Document10 pagesU01 Apps PHSunSMART PHSunSMARTFiles SymbiosysFiles Generated OutputSIPDF 098692O090520182219005228644 1525875549108Jaymar Odtojan AbaloNo ratings yet
- The London Times - "Smallpox Vaccine Triggered AIDS Virus.'"Document29 pagesThe London Times - "Smallpox Vaccine Triggered AIDS Virus.'"compnets_2000@yahoo.ca100% (2)
- 4 +ok+ (11) +Filya+Aulia+Hunou+29-39+doiDocument11 pages4 +ok+ (11) +Filya+Aulia+Hunou+29-39+doiNovy SandhyanaNo ratings yet
- HIV AIDS Opportunistic InfectionsDocument26 pagesHIV AIDS Opportunistic InfectionsLaylee ClareNo ratings yet
- Zimbabwe OSDM Webrevised 2017Document116 pagesZimbabwe OSDM Webrevised 2017Richard Makurumidze Senior0% (1)
- Final ResearchDocument64 pagesFinal Researchhundasa chalaNo ratings yet
- Uts Chapter 2 Lesson 1Document29 pagesUts Chapter 2 Lesson 1John Jerome GironellaNo ratings yet
- Monitoring Equity in Access To Aids Treatment Programmes: A Review of Concepts, Models, Methods and IndicatorsDocument98 pagesMonitoring Equity in Access To Aids Treatment Programmes: A Review of Concepts, Models, Methods and IndicatorsCésar López GodoyNo ratings yet
- UNICEF Annual Report 2019Document68 pagesUNICEF Annual Report 2019Volodymyr DOBROVOLSKYINo ratings yet
- HIV1Document3 pagesHIV1carry123No ratings yet
- HAART PresentationDocument27 pagesHAART PresentationNali peterNo ratings yet
- PMLS TransesDocument2 pagesPMLS TransesjulianneNo ratings yet
- بحث عن مرض الأيدز باللغة الانجليزيةDocument5 pagesبحث عن مرض الأيدز باللغة الانجليزيةdrkamal2No ratings yet
- Construction Health and Safety in South Africa PDFDocument48 pagesConstruction Health and Safety in South Africa PDFGriffiths Ventriloqual ThaumaturgeNo ratings yet